Article Text

Abstract
Background: The acute respiratory distress syndrome (ARDS) is characterised by inflammation of the lung parenchyma and changes in alveolar haemostasis with extravascular fibrin deposition. Factor VII activating protease (FSAP) is a recently described serine protease in plasma and tissues known to be involved in haemostasis, cell proliferation and migration.
Methods: The level of FSAP protein expression was examined by western blotting/ELISA/immunohistochemistry and its activity was investigated by coagulation/fibrinolysis assays in plasma, bronchoalveolar lavage (BAL) fluid and lung tissue of mechanically ventilated patients with early ARDS and compared with patients with cardiogenic pulmonary oedema and healthy controls. Cell culture experiments were performed to assess the influence of different inflammatory stimuli on FSAP expression by various cell populations of the lung.
Results: FSAP protein level and activity were markedly increased in the plasma and BAL fluid of patients with ARDS with a significant contribution to the increased alveolar procoagulant activity. Immunoreactivity for FSAP was observed in alveolar macrophages, bronchial epithelial and endothelial cells of lungs of patients with ARDS, while in controls the immunoreactivity for FSAP was restricted to alveolar macrophages. Only a low basal level of FSAP expression was detected in these cell populations. However, FSAP-specific mRNA expression was induced by lipopolysaccharide and interleukin-8 in human lung microvascular endothelial cells and in bronchial epithelial cells. FSAP was also found to be taken up by alveolar macrophages and degraded within the lysosomal compartment.
Conclusions: Increased levels of FSAP and an altered cellular expression pattern are found in the lungs of patients with ARDS. This may represent a novel pathological mechanism which contributes to pulmonary extravascular fibrin deposition and may also modulate inflammation in the acutely injured lung via haemostasis-independent cellular activities of FSAP.
- ARDS, acute respiratory distress syndrome
- BAL, bronchoalveolar lavage
- FSAP, factor VII activating protease
- IL, interleukin
- LPS, lipopolysaccharide
- TNFα, tumour necrosis factor α
Statistics from Altmetric.com
- ARDS, acute respiratory distress syndrome
- BAL, bronchoalveolar lavage
- FSAP, factor VII activating protease
- IL, interleukin
- LPS, lipopolysaccharide
- TNFα, tumour necrosis factor α
The acute respiratory distress syndrome (ARDS) is characterised by an acute inflammation of the lung parenchyma and is caused by a number of direct (eg, pneumonia) and indirect (eg, sepsis) insults.1,2 The acute phase of ARDS is associated with severe injury of the endothelial and epithelial barrier of the lung, leading to influx of protein-rich oedema fluid into the alveolar and interstitial compartment.1,2 The disturbed integrity of the capillary alveolar barrier and a marked alteration in the alveolar haemostatic balance are the underlying mechanisms for extravascular fibrin deposition in the alveolar space that is characteristic of diverse forms of ARDS.3–5 Procoagulant activity is significantly increased in bronchoalveolar lavage (BAL) fluid of ARDS and fibrinolytic activity is decreased. Tissue factor associated with factor VII and inhibition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1 are major factors responsible for the switch in the alveolar haemostatic balance.3–5 These changes in alveolar haemostasis are observed in extrapulmonary as well as in pulmonary ARDS.3 Tissue fibrin and coagulatory and fibrinolytic intermediates may influence inflammation and lung function in a number of ways—they can increase vascular permeability, influence the expression of inflammatory mediators, and alter the migration and proliferation of inflammatory cells.6–9 Moreover, fibrin and its derivatives are strong inhibitors of surfactant function, contributing to alveolar collapse and gas exchange disturbances in the injured lung.10 In addition, persistent fibrin deposition has been suggested to play a considerable role in the development of post-inflammatory lung fibrosis, providing a matrix on which fibroblasts can migrate and produce collagen.11
On this basis, several animal and clinical studies have investigated the use of anticoagulants or fibrinolytic agents in the treatment of acute lung injury and ARDS. In animals, an improvement in gas exchange, a reduction in inflammation and, at least in some studies, an increased survival have been demonstrated with the administration of anticoagulants such as tissue factor pathway inhibitor,12 site inactivated factor VIIa,13 heparin,14 recombinant hirudin,15 antithrombin16 and activated protein C.17 In addition, aerosolisation of heparin or urokinase18 or overexpression of urokinase-type plasminogen activator in the distal respiratory epithelium19 effectively prevent the development of pulmonary fibrosis following inflammation in bleomycin-induced lung injury. In humans, administration of activated protein C has been shown to improve survival and lung function in patients with severe sepsis.20
Factor VII activating protease (FSAP) is a recently described serine protease in plasma and tissues.21,22 It is produced as a 64 kDa single chain zymogen (scFSAP) which is converted by autoactivation to the proteolytically active two chain form (tcFSAP).23–25 tcFSAP consists of a 46 kDa heavy chain and a 29 kDa active site-bearing light chain connected by a disulfide bridge.23–25 Once generated, tcFSAP is subjected to rapid autodegradation.23–25 FSAP is mainly expressed in the liver, but the protein has been detected in various organs such as lung, kidney, placenta and pancreas.21,26
The precise role of FSAP in different physiological and pathophysiological states is not fully understood. A dual role of FSAP in haemostasis was recently discussed: FSAP is a potent activator of coagulation factor VII, thus promoting in vitro coagulation independently of tissue factor.22 Moreover, FSAP activates pro-urokinase to promote clot lysis in vitro.27 In addition to its role in haemostasis, FSAP—like other haemostatic serine proteases—expresses cellular activities related to cell migration and proliferation. FSAP inhibits the proliferation and migration of vascular smooth muscle cells and endothelial cells in vitro.28,29 Locally applied FSAP was recently shown by our group to be a potent inhibitor of neointimal thickening in a mouse model of wire-induced injury of the femoral artery. This protective effect was mediated by a decrease in cell proliferation and a reduction in the number of vascular smooth muscle cells and inflammatory cells in the neointima, pointing to a possible anti-inflammatory role for FSAP (Sedding et al, unpublished observations).30
To date, FSAP has not been investigated in the context of pulmonary diseases. The objective of the present study was to determine whether FSAP contributes to the changes in alveolar haemostatic balance in the lungs of patients with ARDS, thereby serving as a potential target molecule for therapeutic interventions. In addition, the cellular expression of FSAP under inflammatory conditions was investigated.
METHODS
Study population
All investigational measures were approved by the local ethics committee and written informed consent was obtained from either the patients or their closest relatives.
BAL fluid was obtained by flexible fibreoptic bronchoscopy from 15 spontaneously breathing healthy volunteers without any history of cardiac or lung disease and normal pulmonary function test results (medical students at the Medical School of the Justus-Liebig-University, Giessen, Germany) and from 28 patients. All patients included in this study were recruited from the Intensive Care Unit of the Department of Internal Medicine at the Justus-Liebig-University between 1999 and 2003. The following patient groups were investigated: extrapulmonary ARDS without pulmonary infection (ARDS; n = 15); ARDS with primary lung infection (ARDS + Pneu; n = 8); and cardiogenic pulmonary oedema (CLE; n = 5). Patients fulfilling the inclusion criteria for the different groups as detailed in the online data supplement were included in the study.
All patients required mechanical ventilation. BAL was performed within the first 72 h after the beginning of mechanical ventilation. Arterial oxygen tension/fractional inspired oxygen values, duration of mechanical ventilation, sex, age and smoking history did not differ substantially among the different patient groups. Details on the demographic and clinical data of the patient groups and on the BAL procedure are outlined in the online data supplement.
In addition to BAL fluid, lung specimens from seven patients with ARDS were obtained by autopsy. All patients met the clinical American-European Consensus Conference criteria1 and died in the early phase with a median duration of mechanical ventilation of 92 h. Four patients had ARDS due to pneumonia and three had ARDS of extrapulmonary origin (sepsis). All lung specimens showed the histopathological pattern of diffuse alveolar damage that is characteristic of ARDS. As a control, lung specimens were obtained by autopsy from five individuals who died of myocardial infarction (n = 4) or drug intoxication (n = 1). In each case the pathological conditions of the lung were ruled out by histological examination of lung tissue sections.
FSAP antigen and activity assay
A recently described ELISA technique31 was used to determine the FSAP antigen level in BAL fluid and plasma. FSAP activity was assessed by investigating its single chain urokinase activating potency as recently described31 and by a direct chromogenic assay, as outlined in the online data supplement.
Western blotting
Western blotting for the detection of FSAP was performed using a mixture of two murine monoclonal antibodies directed against light and heavy chain FSAP, respectively. Additional information is provided in the online data supplement.
BAL fluid procoagulant and fibrinolytic activity
The recalcification clotting time of BAL fluid in the absence or presence of an inhibitory antibody against human FSAP was measured using a microcoagulometer. The extent of BAL fluid-induced fibrin clot lysis was determined by a fluorogenic assay as detailed in the online data supplement.
Factor VII activation
Factor VIIa generation in BAL fluid in the absence or presence of an inhibitory antibody against human FSAP was assessed with a factor VIIa-specific chromogenic substrate as outlined in the online data supplement.
Immunohistochemistry
Immunohistochemistry for the detection of FSAP in formalin-fixed paraffin-embedded lung tissue was performed using Histostain-SP Kit according to the manufacturer’s instructions (Zymed Laboratories Inc, San Francisco, California, USA) with the same mixture of antibodies against FSAP as described for the western blot experiments. Controls were performed by substituting the primary by a non-specific antibody. For safe identification of FSAP positive cells, immunohistochemical staining was performed on serial sections using antibodies directed against CD68 (alveolar macrophages), von Willebrand factor (endothelial cells), vimentin (fibroblasts) and pro-surfactant protein C (alveolar type II cells). Details are outlined in the online data supplement.
Cell culture
Lung microvascular endothelial cells were purchased from Clonetics (San Diego, California, USA). Human primary bronchial airway epithelial cells were isolated from non-used donor lungs without a history of pulmonary disease at the time of lung transplantation as recently described.32 BAL fluid alveolar macrophages from healthy volunteers were purified by adherence to plastic tissue culture dishes as recently described.33 Details on the cell culture conditions are provided in the online data supplement.
Cell stimulation, RNA isolation and reverse transcriptase (RT) reaction
Subcultures of lung microvascular endothelial cells, primary bronchial airway epithelial cells and alveolar macrophages were either unstimulated or stimulated with various concentrations of lipopolysaccharide (LPS) from Escherichia coli (0.01–1 μg/ml) for 4 h or with 0.5 μg/ml LPS for 2–12 h. Furthermore, lung microvascular endothelial cells were stimulated for 2–12 h with interleukin (IL)-6 (10 ng/ml), tumour necrosis factor (TNF)α (20 ng/ml), IL-8 (25 ng/ml) and IL-1β (5 ng/ml), respectively, or for 8 h with IL-8 (25 ng/ml) or LPS (0.5 μg/ml) in the absence or presence (1 μg/ml) of an anti-IL-8 antibody. Total cellular RNA was extracted using QIAzol lysis reagent (Qiagen, Hilden, Germany) and 1 μg total RNA was reverse transcribed as detailed in the online data supplement.
Relative FSAP mRNA quantification by real-time PCR
The regulation of FSAP mRNA expression in stimulated cells was analysed by real-time quantitative PCR using the ΔΔCT method for the calculation of relative changes.34 Real-time PCR was performed by the Sequence Detection System 7700 (PE Applied Biosystems) as outlined in the online data supplement. β-actin was used as reference.
Uptake of FSAP by alveolar macrophages
Mouse alveolar macrophages were pretreated with 70 nM LysoTracker (Cambrex Bio Science, Walkersville, Massachusetts, USA) and subsequently incubated with 2 µg/ml human FSAP for 10, 30 or 60 min followed by immuno-staining and western blot analysis for the detection of FSAP at each time point, as described in the online data supplement. In some experiments, cells were preincubated with 100 µM chloroquine 2 h before the addition of human FSAP.
Analysis of data
The statistical analyses were performed in R Version 2.3.1.35 Deviations from the normal distribution were tested using the Shapiro-Wilk test. All in vitro data were normally distributed so these data are presented as mean (SD). Clinical data are given as median and interquartile range. The box-and-whisker-plots indicate the median, 1st and 3rd quartile; the whiskers are extended to the most extreme value inside the 1.5-fold interquartile range. Differences between two groups were tested with the Student t test and Wilcoxon rank sum test according to the distribution of the data. All tests were performed with an undirected hypothesis (two-sided). The level of statistical significance was set at 5%.
RESULTS
ELISA experiments revealed significantly raised FSAP antigen levels in the BAL fluid of patients with extrapulmonary ARDS compared with healthy controls. Accordingly, FSAP activity in the BAL fluid of these patients was found to be significantly increased, regardless of whether FSAP activity was assessed by its single chain urokinase activating potency (fig 1) or by a direct chromogenic assay (see fig 1 in the online data supplement available at http://thorax.bmj.com/supplemental). In addition, a significant increase in FSAP antigen level and activity in the plasma of these patients was observed. FSAP antigen level and activity were also raised in the BAL fluid of patients with pulmonary ARDS, but to a lesser extent. Only a slight increase in FSAP antigen and activity were noted in the plasma of this patient group. No significant change in FSAP antigen level and activity were found in the BAL fluid or plasma of patients with cardiogenic pulmonary oedema (fig 1). In line with these observations, western blot experiments detected significant amounts of the 46 kDa heavy chain and the 29 kDa light chain of the proteolytically active two chain form of FSAP in plasma and BAL fluid of patients with ARDS but not in patients with cardiogenic pulmonary oedema or healthy controls (fig 2A and B). For detecting the 29 kDa light chain in BAL fluid from patients with ARDS, the BAL fluid was concentrated before western blotting (inset of fig 2B). Increased levels of the heavy and light chain of the active two chain form of FSAP were also detected in the lung tissue of patients with ARDS (fig 2C). Interestingly, in contrast to BAL fluid, heavy and light chain FSAP was detected in comparable quantities, but no single chain FSAP was detectable in the lung tissue. A possible explanation is that two chain FSAP may be stabilised when bound to the cell surface and/or extracellular matrix. Accordingly, we have observed strong binding of two chain FSAP but not single chain FSAP to the extracellular matrix protein vitronectin (Wygrecka et al, unpublished observations).

Quantitation of factor VII activating protease (FSAP) antigen and activity in bronchoalveolar lavage fluid (BALF) and plasma of patients with acute respiratory distress syndrome (ARDS) compared with healthy controls and patients with cardiogenic pulmonary oedema. FSAP antigen level as assessed by ELISA (top panels) and FSAP activity as assessed by its single chain urokinase activating potency (bottom panels) in BAL fluid (left) and plasma (right) of healthy controls (n = 15) and of patients with extrapulmonary ARDS without pulmonary infection (ARDS; n = 15), ARDS with primary lung infection (ARDS + Pneu; n = 8) and with cardiogenic pulmonary oedema (CLE; n = 5) were quantitated. The box-and-whisker plots indicate the median, 1st and 3rd quartiles; the whiskers are extended to the most extreme value inside the 1.5-fold interquartile range. *p = 0.016, **p = 0.009, ***p<0.001 ARDS vs healthy controls.

Western blot analysis of factor VII activating protease (FSAP) protein in plasma, bronchoalveolar lavage (BAL) fluid and lung tissue. Western blotting with FSAP specific antibodies was performed to characterise FSAP protein in (A) plasma and (B) BAL fluid of healthy controls and of patients with extrapulmonary acute respiratory distress syndrome (ARDS) without pulmonary infection (ARDS), ARDS with primary lung infection (ARDS + Pneu) and with cardiogenic pulmonary oedema (CLE) as indicated. Representative patients for each entity are shown (healthy controls: 2/15, CLE: 2/5, ARDS: 3/15; ARDS + Pneu: 2/8). The inset shows the presence of the 29 kDa light chain of FSAP in concentrated BAL fluid from patients with ARDS but not from controls (healthy controls: 3/3, ARDS: 3/3). (C) Characterisation of FSAP protein in lung homogenate of patients with ARDS and controls (shown: myocardial infarction). Representative patients for each entity are shown (controls: 3/5, ARDS: 3/3; ARDS + Pneu: 3/4). scFSAP, single chain FSAP; hcFSAP, heavy chain of proteolytically active two chain FSAP; lcFSAP, light chain of proteolytically active two chain FSAP.
FSAP has a dual role in haemostasis, being involved in both procoagulant and fibrinolytic pathways. We therefore investigated both these properties in BAL fluid from patients with ARDS and healthy controls. We observed a significant contribution of FSAP to the increased procoagulant activity in the BAL fluid of patients with ARDS, as evidenced by a prolonged clotting time of these samples in the presence of a neutralising antibody against FSAP (fig 3A). Since the neutralising anti-FSAP antibody may also be directed against activated FSAP in the standard human plasma used in these experiments, the coagulation assay has also been performed using FSAP-deficient plasma. However, clotting times were not significantly altered using FSAP-deficient instead of standard human plasma (fig 3A). In contrast to the contribution of FSAP to increased procoagulant activity, FSAP did not significantly alter fibrinolytic activity of the BAL fluid in patients with ARDS (fig 3B). To further support a potential role for FSAP in coagulation processes in BAL fluid from patients with ARDS, we investigated the activation of factor VII in BAL fluid from these patients in the absence or presence of an inhibitory antibody directed against human FSAP. Factor VII activation in BAL fluid from patients with ARDS was significantly reduced by the anti-FSAP antibody (fig 4). Taken together, these data indicate that FSAP contributes to procoagulant activity in the BAL fluid of patients with ARDS via generation of factor VIIa.

Analysis of factor VII activating protease (FSAP) procoagulant and fibrinolytic activity in bronchoalveolar lavage (BAL) fluid of patients with acute respiratory distress syndrome (ARDS). (A) The procoagulant activity of BAL fluid from patients with ARDS was assessed by plasma clotting assay in the absence or presence of an inhibitory antibody against FSAP (anti-FSAP) or a control antibody (IgG) using standard human plasma (grey boxes) or FSAP-deficient plasma (white boxes). (B) BAL fluid-induced lysis of fibrin clots was assessed by means of a fluorogenic assay and is given for patients with ARDS in the absence or presence of an inhibitory antibody against FSAP (anti-FSAP) or a control antibody (IgG) in relation to healthy controls. The box-and-whisker plots indicate the median, 1st and 3rd quartile; the whiskers are extended to the most extreme value inside the 1.5-fold interquartile range (n = 10). *p<0.001 ARDS vs ARDS + anti-FSAP antibody.
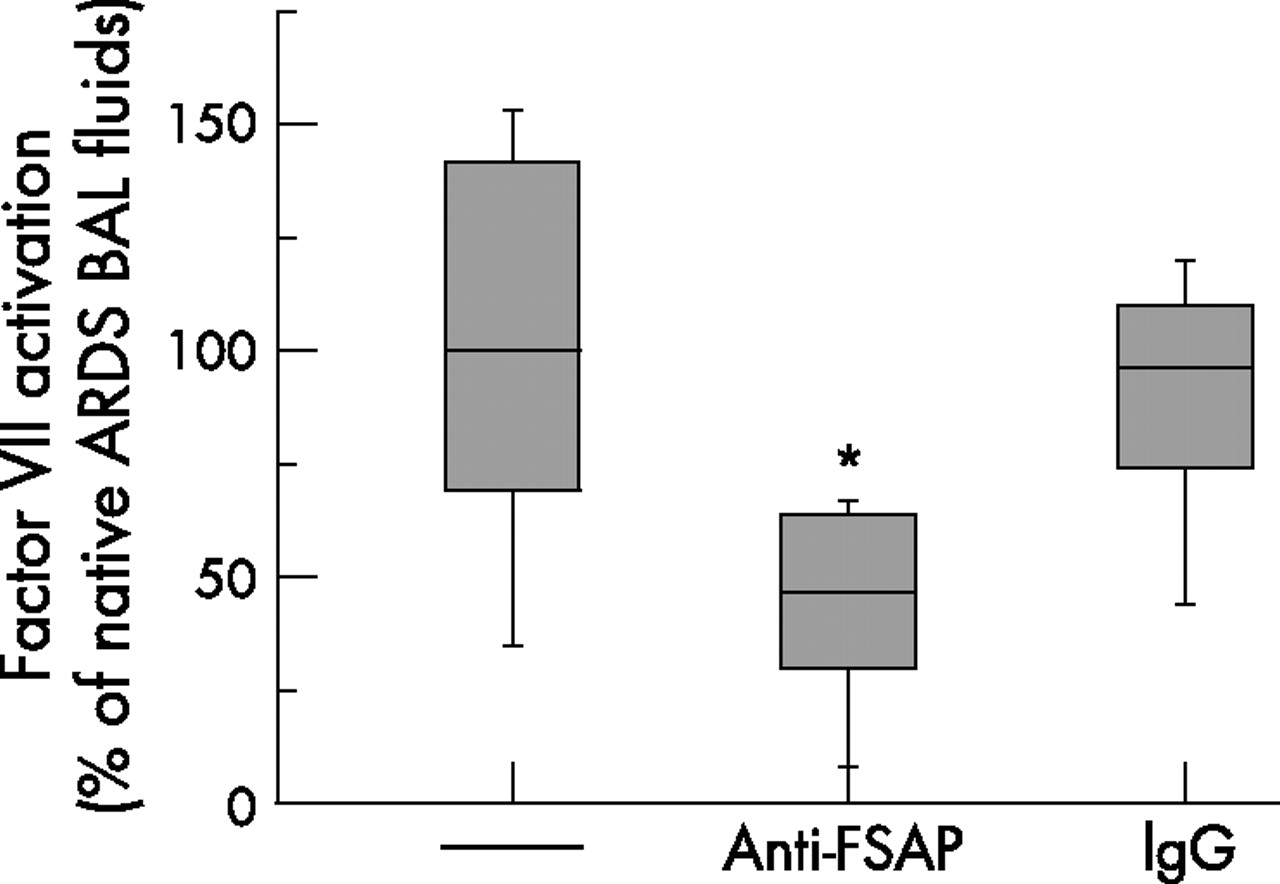
Factor VII activation in bronchoalveolar lavage (BAL) fluid from patients with acute respiratory distress syndrome (ARDS). Factor VII activation in BAL fluid from patients with ARDS was assessed by means of a factor VIIa specific chromogenic substrate assay in the absence or presence of an inhibitory antibody directed against human factor VII activating protease (FSAP) or a control antibody (IgG). Data are presented in relation to BAL fluid from patients with ARDS in the absence of antibodies (100%). The box-and-whisker plots indicate the median, 1st and 3rd quartile; the whiskers are extended to the most extreme value inside the 1.5-fold interquartile range (n = 10). *p = 0.03 for ARDS vs ARDS + anti-FSAP antibody.
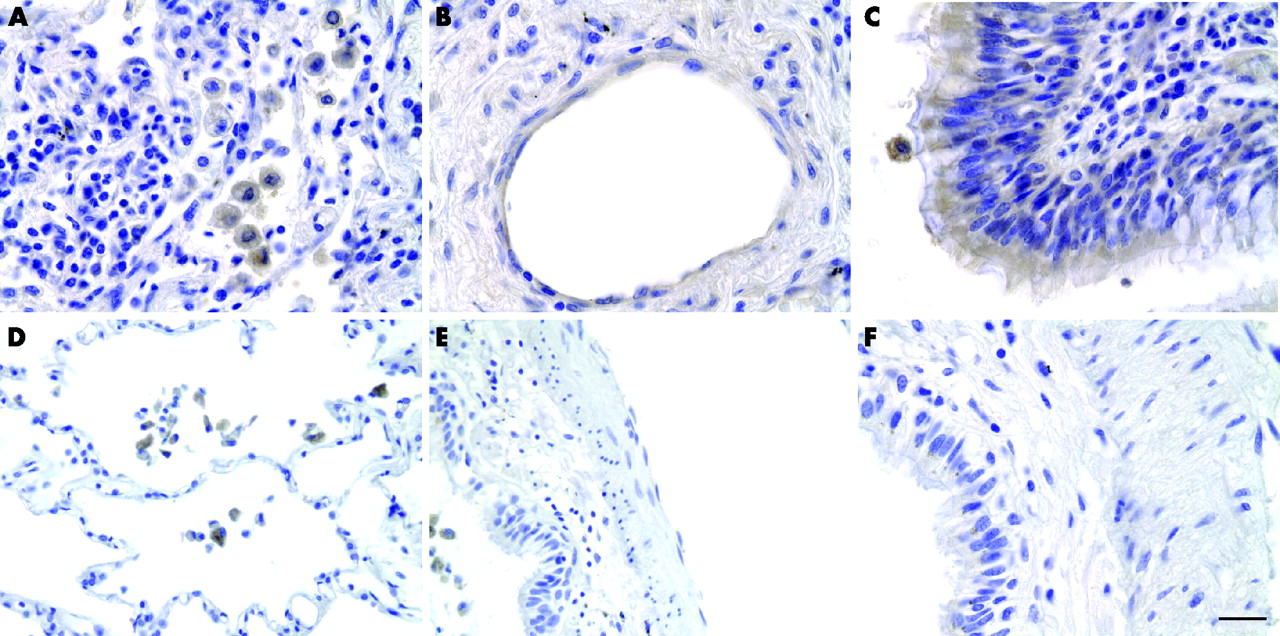
Immunohistochemical studies showed a wider distribution of FSAP in the lungs of patients with ARDS than in control lungs (fig 5). FSAP positive staining in control lungs was restricted to alveolar macrophages (fig 5D–F) while, in the lungs of patients with ARDS, FSAP was present in macrophages, endothelial and bronchial epithelial cells (fig 5A–C). FSAP was also detected in alveolar macrophages of the BAL fluid of subjects with ARDS and healthy controls (not shown).

Localisation of factor VII activating protease (FSAP) in lung tissue. Sections from lung tissue of patients with acute respiratory distress syndrome (ARDS) (A–C) or controls (myocardial infarction; D–F) were stained for FSAP with specific antibodies (brown colour). In ARDS, strong immunoreactivity for FSAP was observed in (A) alveolar macrophages, (B) endothelial cells and (C) bronchial epithelial cells. In controls FSAP protein was detected in alveolar macrophages only (D) and not in other cells such as (E) endothelial or (F) bronchial epithelial cells. One representative patient out of seven with ARDS and five controls is shown. Bar = 5 μm.
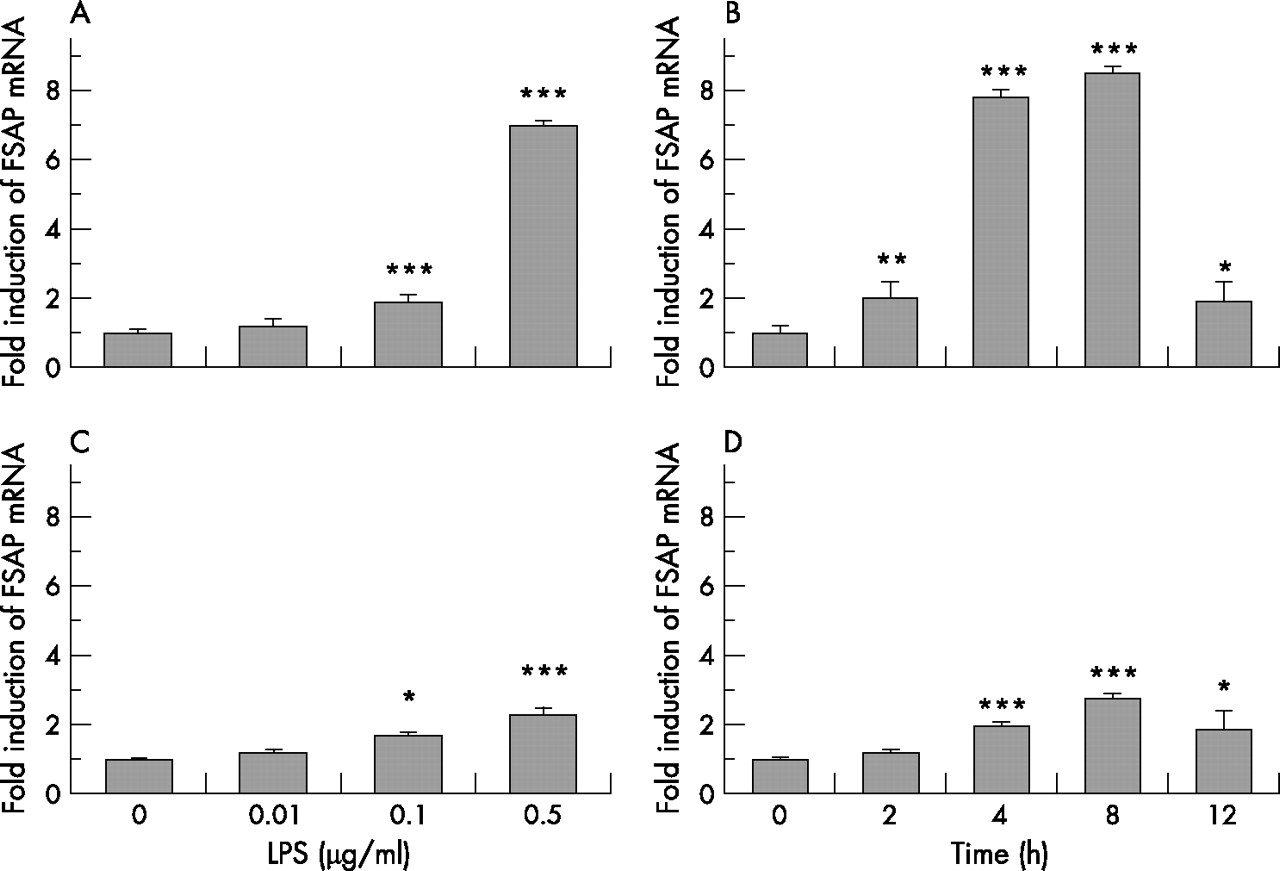
To further characterise the cellular expression pattern of FSAP under inflammatory conditions, cells that stained positive for FSAP in the lungs of patients with ARDS were stimulated with LPS and the expression of FSAP-specific mRNA in these cells was assessed by real-time RT-PCR. Only a low basal expression of FSAP was observed in these cell populations but, in lung microvascular endothelial cells as well as in bronchial airway epithelial cells, FSAP expression was inducible by LPS in a dose-dependent and time-dependent manner. In endothelial cells the effect of LPS on FSAP mRNA expression was dose-dependent over the range 0.01–0.5 μg/ml after 4 h of treatment (fig 6A). Higher doses of LPS resulted in significant cell death as assessed by lactate dehydrogenase cytotoxicity assay (not shown). Maximal stimulation was seen at 0.5 μg/ml (sevenfold increase in FSAP mRNA expression compared with unstimulated control; fig 6A). This concentration was chosen to investigate the time-dependent expression of FSAP mRNA in these cells. The expression of FSAP mRNA was doubled after 2 h of treatment and the maximal effect was seen at 8 h (8.5-fold increase; fig 6B). Bronchial epithelial cells were less sensitive than endothelial cells to LPS stimulation. Maximal stimulation was seen at 0.5 μg/ml LPS after 8 h of treatment (2.8-fold increase; figs 6C and D). Once again, higher doses of LPS resulted in significant cell death.

Dose-response and time course induction of factor VII activating protease (FSAP) mRNA by lipopolysaccharide (LPS) in lung microvascular endothelial cells (A, B) and bronchial airway epithelial cells (C, D). The cells were stimulated for 4 h with 0.01–0.5 μg/ml LPS (A and C) or treated with 0.5 μg/ml LPS for 2–12 h (B and D). Total cellular RNA was isolated and reverse-transcribed into cDNA, and cDNA was analysed for the mRNA expression of FSAP and β-actin by real-time PCR. Results are expressed as a ratio of target gene to β-actin mRNA control and are mean (SD) values for relative FSAP mRNA levels from five independent experiments. *p = 0.03 (endothelial cells after 12 h treatment), *p = 0.04 (bronchial airway epithelial cells after treatment with 0.1 μg/ml LPS and after 12 h treatment); **p = 0.007; ***p<0.001; all versus non-treated cells.
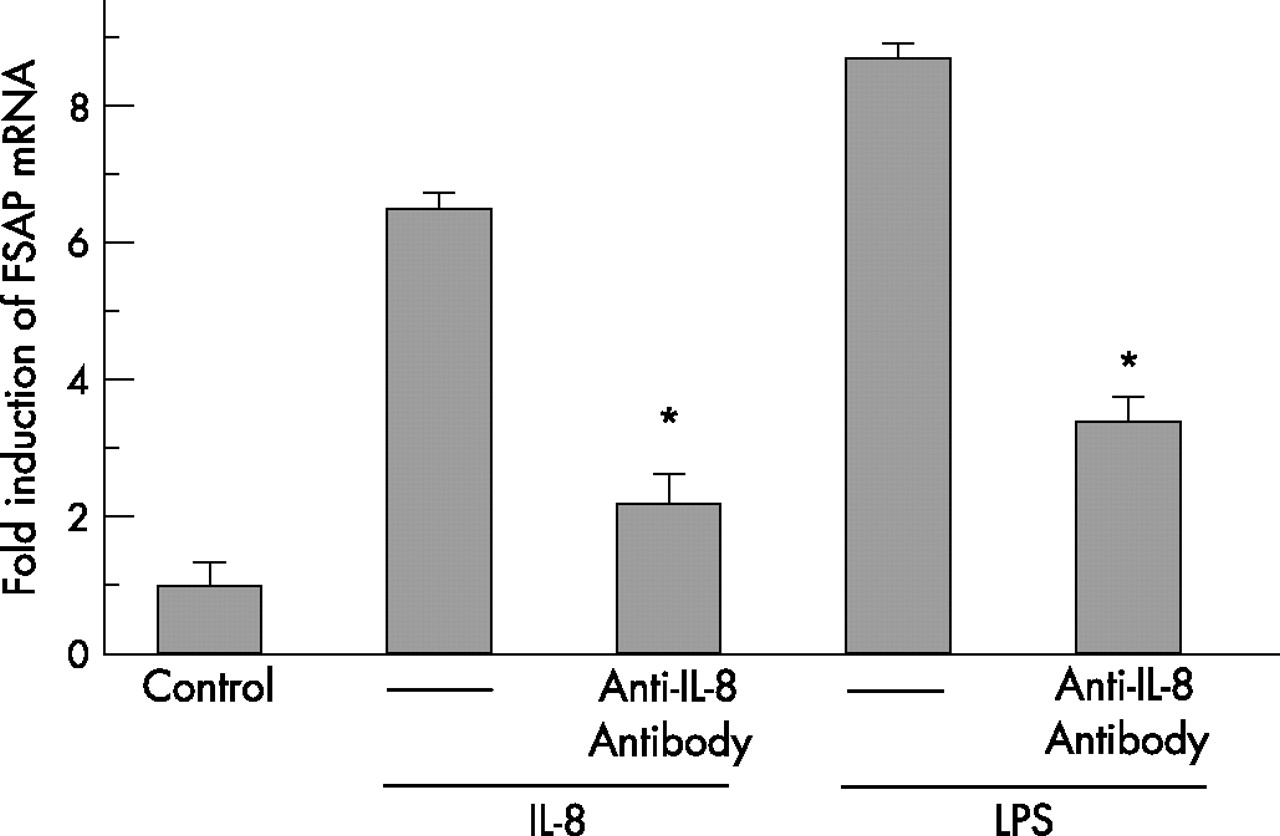
Since the strongest induction of FSAP mRNA expression was observed in lung microvascular endothelial cells, we further examined FSAP mRNA expression in this cell type after treatment with various inflammatory mediators. No induction of FSAP expression was detected after IL-1β stimulation (fig 7D). FSAP production was slightly increased by IL-6 (threefold at 4 h; fig 7A) and TNFα (twofold at 2 h; fig 7B) but IL-8 strongly upregulated the FSAP mRNA level (sevenfold at 8 h; fig 7C). We also found that LPS induced expression of FSAP in lung microvascular endothelial cells was markedly inhibited by an anti-IL-8 antibody, indicating that LPS-dependent FSAP production in this cell type is at least partially mediated by endogenously produced IL-8 (fig 8).
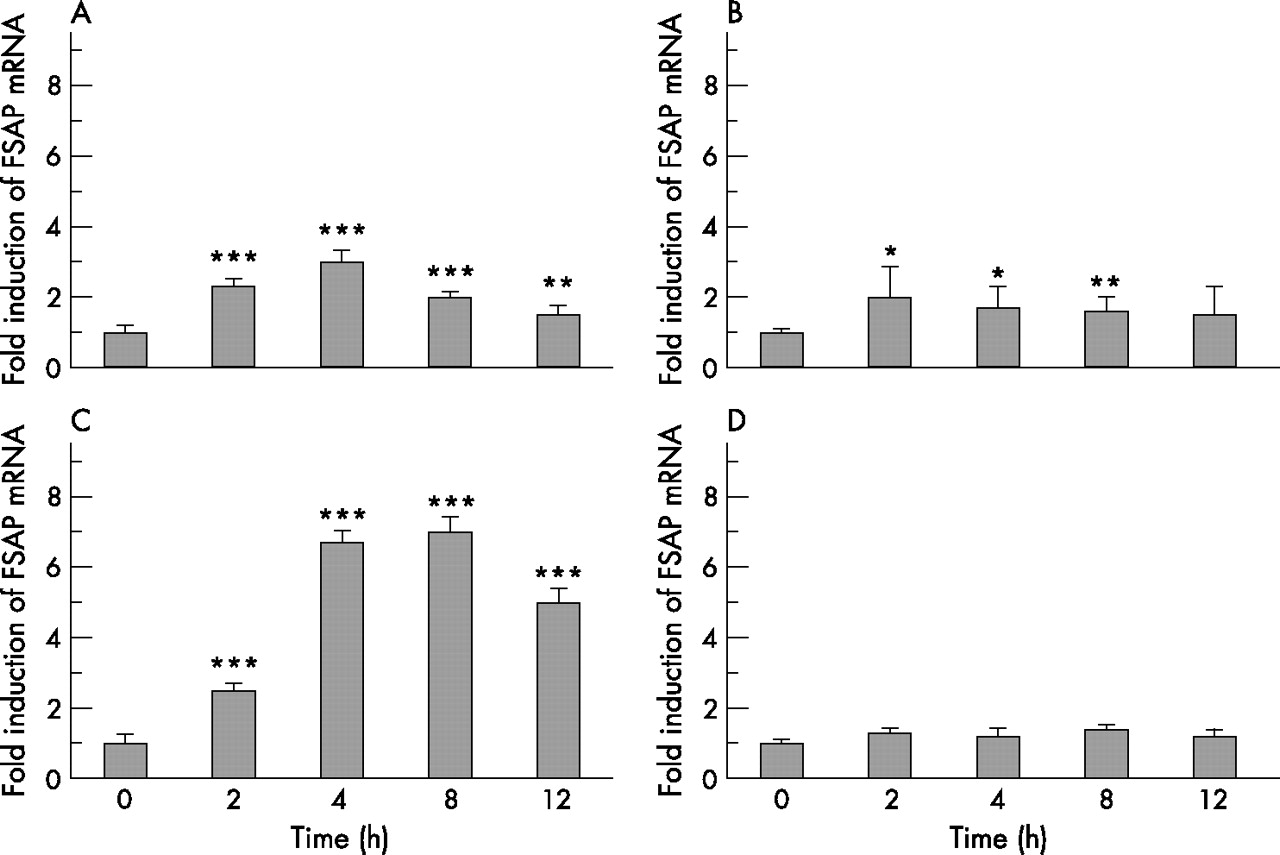
Time course induction of factor VII activating protease (FSAP) mRNA by interleukin (IL)-6, tumour necrosis factor (TNF)α, IL-8 and IL-1β in lung microvascular endothelial cells. Lung microvascular endothelial cells were stimulated for 2–12 h with (A) 10 ng/ml IL-6, (B) 20 ng/ml TNFα, (C) 25 ng/ml IL-8 and (D) 5 ng/ml IL-1β. Total cellular RNA was isolated and reverse-transcribed into cDNA. cDNA was analysed for the mRNA expression of FSAP and β-actin by real-time PCR. Results are expressed as a ratio of target gene to β-actin mRNA control and are mean (SD) values for relative FSAP mRNA levels from five independent experiments. *p = 0.03, **p = 0.02, ***p<0.001, stimulated vs non-stimulated cells.

Factor VII activating protease (FSAP) mRNA expression by lung microvascular endothelial cells after stimulation with either interleukin (IL)-8 or lipopolysaccharide (LPS) in the presence or absence of an anti-IL-8 antibody. Lung microvascular endothelial cells were stimulated for 8 h with either 25 ng/ml IL-8 or 0.5 μg/ml LPS in the presence or absence of an anti-IL-8 antibody. Total cellular RNA was isolated and reverse-transcribed into cDNA. cDNA was analysed for the mRNA expression of FSAP and β-actin by real-time PCR. Results are expressed as a ratio of target gene to β-actin mRNA control and are mean (SD) values for relative FSAP mRNA levels from five independent experiments. *p<0.001, LPS vs LPS + anti-IL-8 antibody and IL-8 vs IL-8 + anti-IL-8 antibody.
In contrast to endothelial and bronchial epithelial cells, FSAP mRNA expression was not inducible in alveolar macrophages by any of the inflammatory mediators (not shown). This observation led us to propose that the strong immunoreactivity for FSAP in alveolar macrophages is due to uptake and degradation of FSAP rather than to FSAP expression. The uptake of human FSAP by cultured mouse alveolar macrophages was subsequently investigated. After incubation for 10 and 30 min, human FSAP was detected in mouse alveolar macrophages as assessed by immunocytochemistry (fig 9B) and by western blotting of the macrophage cell extracts (fig 9A), but a time-dependent loss of human FSAP in mouse alveolar macrophages was noted. Furthermore, co-localisation of human FSAP and lysosomes in the cytoplasm of mouse alveolar macrophages revealed that FSAP was directed to the lysosomal compartment (fig 9B). Addition of 100 µM chloroquine to the medium prevented the time-dependent loss of human FSAP in mouse alveolar macrophages by blocking lysosomal degradation (fig 9A). These data indicate that FSAP is taken up by alveolar macrophages and degraded in the lysosomal compartment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Uptake of factor VII activating protease (FSAP) by alveolar macrophages. (A) After incubation of mouse alveolar macrophages with human FSAP in the absence (left panel) or the presence of chloroquine (right panel) for 10, 30 and 60 min, respectively, cell extracts were analysed by western blotting using an antibody specific for human FSAP. In parallel, cell extracts were analysed in the absence of FSAP (control = Co); hcFSAP, heavy chain of proteolytically active two chain FSAP; lcFSAP, light chain of proteolytically active two chain FSAP. (B) At the indicated time points, cells were stained for human FSAP (left panels) or for lysosomes (middle panels) and an overlay of both images was generated in each case (right panels). Bar = 5 μm. One representative experiment out of five independent experiments for each time point is shown. The insets show high power images of single macrophages.
DISCUSSION
FSAP is a recently described serine protease in plasma and tissues with a potential role in haemostasis, cell proliferation and inflammation.21–30 In the present study we found markedly increased FSAP protein levels and activity in the plasma and lungs of patients with ARDS. FSAP was found to contribute to factor VIIa generation and to increased procoagulant activity in the BAL fluid of patients with ARDS. Furthermore, a differential cellular distribution pattern of FSAP in the lung tissue of patients with ARDS and healthy controls was observed, whereby FSAP protein was detected in alveolar macrophages, bronchial epithelial and endothelial cells of the lungs of patients with ARDS. FSAP expression in the latter cell types was found to be inducible by LPS and IL-8. Moreover, we identified alveolar macrophages to be centrally involved in FSAP metabolism in the lung, as they internalise FSAP followed by lysosomal degradation.
Changes in the FSAP level and activity in the BAL fluid of patients with ARDS were compared with patients with cardiogenic pulmonary oedema in the absence of ARDS and lung infection. The duration of mechanical ventilation was comparable (52 vs 60 h) and there was a similar disturbance in gas exchange, as evidenced by similar arterial oxygen tension to fractional inspired oxygen ratios (184 vs 201 mm Hg). No significant change in FSAP level and activity in the BAL fluid of patients with cardiogenic pulmonary oedema was observed compared with healthy controls. These findings indicate that it is not the mechanical ventilation per se that is responsible for the observed changes in FSAP level and activity in the lungs of patients with ARDS. Interestingly, virtually identical changes in FSAP level and activity were observed in the lungs of patients with extrapulmonary ARDS without pulmonary infection and in those with ARDS with primary lung infection. This is in line with a previous study showing that altered levels of coagulatory and fibrinolytic intermediates can be observed in the BAL fluid of patients with acute inflammatory lung diseases, whether triggered by extrapulmonary systemic events or primary lung infection.3
The origin of increased FSAP level and activity in the lungs of patients with ARDS is not currently known. FSAP has previously been shown to be mainly expressed in the liver.21,26 Increased production of FSAP in the liver with subsequent (auto)-activation of FSAP may largely account for increased plasma levels of proteolytically active FSAP as observed in ARDS of extrapulmonary origin. Increased endothelial and epithelial permeability may then favour leakage of proteolytically active FSAP from the plasma into the alveolar compartment. Extracellular RNA was recently shown to serve as a negatively charged surface to promote the (auto)-activation of FSAP36 and may therefore be involved in the (auto)-activation of FSAP in the systemic circulation of patients with ARDS. Accordingly, raised RNA levels have recently been observed in the plasma of subjects with ARDS (Wygrecka et al, unpublished observations). In contrast, only a slight upregulation of proteolytically active FSAP was observed in the systemic circulation of ARDS induced by primary lung infection. In this patient entity as well as in extrapulmonary ARDS, leakage of the single chain zymogen from the blood into the alveolar space with subsequent activation to the proteolytically active two chain form in the alveoli may therefore also contribute to increased FSAP levels and activity in the lungs of patients with ARDS. Moreover, our data indicate that the lung itself—particularly endothelial and bronchial epithelial cells—may also represent a source of FSAP in ARDS.
Another important finding of our study is the observation that alveolar macrophages appear to be centrally involved in the clearance of FSAP from the lung. These investigations were driven by the finding that healthy controls, as well as patients with ARDS, showed strong staining for FSAP protein in alveolar macrophages despite very minor basal and inducible FSAP-specific mRNA expression in these cells. Our data evidently indicate that FSAP is taken up by alveolar macrophages and degraded within the lysosomal compartment. The detailed mechanism of FSAP uptake into alveolar macrophages needs to be clarified in further investigations. We recently observed complex formation between FSAP and plasminogen activator inhibitor-1 in the BAL fluid of patients with ARDS and in a purified system (Wygrecka et al, unpublished observations). Since complexes of plasminogen activator inhibitor-1 with other serine proteases such as urokinase are internalised into cells via the low density lipoprotein receptor-related protein with subsequent degradation in lysososmes,37 this might be a possible mode of FSAP internalisation into alveolar macrophages as well.
Increased levels of procoagulant FSAP in the lungs of patients with ARDS, as observed in the present study, may represent a novel pathological mechanism which contributes to the progression of ARDS. In line with these considerations, FSAP activity in BAL fluid from patients with ARDS was positively correlated with the Acute Physiology and Chronic Health Evaluation (APACHE) II score, although the correlation did not reach statistical significance (see fig 2 in the data supplement available online at http://thorax.bmj.com/supplemental). An increased alveolar procoagulant activity and persistent alveolar fibrin deposition are believed to contribute to the impairment of gas exchange and to the induction of post-inflammatory fibroproliferative processes in the lungs of patients with ARDS.10,11,38 In previous reports, tissue factor and factor VII had been identified as major contributors to the increased procoagulant activity in the BAL fluid of patients with ARDS.3–5 The current data indicate that at least a part of the extrinsic coagulation pathway activation observed in the alveolar space of patients with ARDS may be triggered by FSAP. It therefore seems reasonable to propose a potential contribution of FSAP to pulmonary fibrin deposition in ARDS, thereby serving as a potential target molecule for therapeutic interventions. On the other hand, FSAP expresses haemostasis-independent activities related to cell migration and proliferation, and a possible anti-inflammatory role for FSAP has recently been proposed via inhibition of inflammatory cell proliferation and migration. These activities are mediated by its interference with different growth factors and/or by its ability to activate pro-urokinase which is significantly enhanced in the presence of polyanions that can be found on cell surfaces and associated with the extracelullar matrix.27–29 In this context, it was recently demonstrated that the topical application of FSAP can inhibit wire injury-induced neointimal lesion formation in the mouse. This protective effect was mediated by its inhibitory effect on the proliferation and migration of vascular smooth muscle cells and monocytes/macrophages with a significant reduction in the number of inflammatory cells in the neointima of the FSAP-treated vessels (Sedding et al, unpublished observations).30 Keeping in mind these observations, it is easy to imagine that increased levels and an altered expression of FSAP, as observed in the present study, may modulate inflammation in the lungs of patients with ARDS via the haemostasis-independent cellular activities. In this way, FSAP may even exert a beneficial effect in the acutely injured lung. Exogenous administration and blockage of FSAP, respectively, in animal models of lung injury will further clarify whether FSAP has a critical role in ARDS and whether it is harmful or protective.
We conclude that increased levels and an altered cellular expression pattern of FSAP is observed in the lungs of patients with ARDS. This may represent a novel pathological mechanism which contributes to changes in alveolar haemostasis and to extravascular fibrin deposition in the lungs of patients with ARDS, suggesting that FSAP may serve as a potential target molecule for therapeutic interventions. In addition, this may modulate inflammation in the acutely injured lung via haemostasis-independent cellular activities of FSAP.
Acknowledgments
The authors thank Gisela Mueller and Horst Thiele for excellent technical assistance and Dr Sandip M Kanse for providing purified human FSAP for the cell culture experiments.
REFERENCES
Supplementary materials
Files in this Data Supplement:
Footnotes
-
Published Online First 3 May 2007
-
This study was supported by Deutsche Forschungsgemeinschaft (DFG; SFB 547, KFO 118).
-
Competing interests: None.
-
MW and PM contributed equally to the manuscript.