Article Text

Abstract
Background: Mast cell recruitment and activation are critical for the initiation and progression of inflammation and fibrosis. Mast cells infiltrate specific structures in many diseased tissues such as the airway smooth muscle (ASM) in asthma. This microlocalisation of mast cells is likely to be key to disease pathogenesis. Human lung mast cells (HLMC) express the Ca2+ activated K+ channel KCa3.1 which modulates mediator release, and is proposed to facilitate the retraction of the cell body during migration of several cell types. A study was undertaken to test the hypothesis that blockade of KCa3.1 would attenuate HLMC proliferation and migration.
Methods: HLMC were isolated and purified from lung material resected for bronchial carcinoma. HLMC proliferation was assessed by cell counts at various time points following drug exposure. HLMC chemotaxis was assayed using standard Transwell chambers (8 μm pore size). Ion currents were measured using the single cell patch clamp technique.
Results: KCa3.1 blockade with triarylmethane-34 (TRAM-34) did not inhibit HLMC proliferation and clotrimazole had cytotoxic effects. In contrast, HLMC migration towards the chemokine CXCL10, the chemoattractant stem cell factor, and the supernatants from tumour necrosis factor α stimulated asthmatic ASM was markedly inhibited with both the non-selective KCa3.1 blocker charybdotoxin and the highly specific KCa3.1 blocker TRAM-34 in a dose dependent manner. Although KCa3.1 blockade inhibits HLMC migration, KCa3.1 is not opened by the chemotactic stimulus, suggesting that it must be involved downstream of the initial receptor-ligand interactions.
Conclusions: Since modulation of KCa3.1 can inhibit HLMC chemotaxis to diverse chemoattractants, the use of KCa3.1 blockers such as TRAM-34 could provide new therapeutic strategies for mast cell mediated diseases such as asthma.
- ASM, airway smooth muscle
- HLMC, human lung mast cell
- IL, interleukin
- SCF, stem cell factor
- TGF-β, transforming growth factor β
- TNF-α, tumour necrosis factor α
- TRAM-34, triarylmethane-34
- mast cell
- migration
- asthma
- CXCL10
- KCa3.1
Statistics from Altmetric.com
- ASM, airway smooth muscle
- HLMC, human lung mast cell
- IL, interleukin
- SCF, stem cell factor
- TGF-β, transforming growth factor β
- TNF-α, tumour necrosis factor α
- TRAM-34, triarylmethane-34
Mast cells are major effector cells in many different inflammatory and fibrotic diseases including asthma, pulmonary fibrosis, and rheumatoid arthritis.1 In these diseases there is evidence of increased mast cell numbers within specific tissue structures,2,3 where they contribute to the immunopathology through the release of a plethora of pleiotropic autacoids, proteases, and cytokines.1 For example, in asthma, mast cells infiltrate the airway smooth muscle (ASM) bundles in the airways of asthmatic subjects, but not those of either patients with eosinophilic bronchitis or normal subjects.2 This event is likely to be key in the development of the disordered ASM physiology of asthma. Similarly, in rheumatoid disease there is accumulation of mast cells in the pannus at the edge of articular cartilage erosion.3 It follows from this that targeting mast cell migration to these sites may offer a novel means of treating diseases in which mast cells play a role.
We have recently shown that ASM cell derived CXCL10 (also known as interferon-γ inducible protein of 10 kDa (IP-10)) induces human lung mast cell (HLMC) chemotaxis.4 CXCL10 induces HLMC migration by binding to the chemokine receptor CXCR3 which is preferentially expressed on mast cells within the ASM of asthmatic subjects.5 Inhibition of the CXCL10/CXCR3 axis could therefore provide a new and effective treatment for asthma. However, in addition to CXCL10, the ASM produces several other mast cell chemoattractants including stem cell factor (SCF),6 transforming growth factor β (TGFβ),7 and the chemokines CXCL8, CXCL12 and CCL11.5 A more general approach to preventing mast cell migration may therefore demonstrate better efficacy in the treatment of asthma and other mast cell mediated diseases than targeting of specific chemokines.
Ion channels are emerging as interesting therapeutic targets in both inflammatory and structural non-excitable cells.8,9 HLMC, human bone marrow derived and human peripheral blood derived mast cells express the intermediate conductance Ca2+ activated K+ channel, KCa3.1 (also known as IKCa1).10–,12 This channel maintains a negative membrane potential during cell activation, thus increasing the driving potential for Ca2+ influx through store operated calcium channels.13 Blockade of KCa3.1 therefore attenuates HLMC degranulation while opening it enhances it.10,11 In addition to their role in cell activation, ion channels carrying K+ and Cl− have been implicated in many diverse cellular processes including proliferation, chemotaxis, and apoptosis. KCa channels are required for lysophosphatidic acid induced microglial cell migration14 and evidence also exists for the involvement of KCa3.1 in both T and B cell proliferation.15
In this study we have examined the hypothesis that KCa3.1 is important for HLMC migration and proliferation. To test this we examined the effects of KCa3.1 blockade with the relatively non-selective KCa3.1 blocker charybdotoxin and the highly specific KCa3.1 blocker triarylmethane-34 (TRAM-34) on HLMC migration induced by CXCL10, SCF, and ASM. We also studied the effect of the KCa3.1 blockers TRAM-34 and clotrimazole on HLMC proliferation.
METHODS
Reagents
The following were purchased: recombinant human (rh)SCF, rhIL-6, rhIL-10, rhCXCL10 (R&D, Abingdon, UK); 1-ethyl-2-benzimidazolinone (1-EBIO), charybdotoxin, iberiotoxin (Sigma, Poole, Dorset, UK); mouse IgG1 mAb YB5.B8 (anti-CD117) (Cambridge Bioscience, Cambridge, UK); sheep anti-mouse IgG1 Dynabeads (Dynal, Wirral, UK); DMEM/Glutamax/Hepes, antibiotic/antimycotic solution, MEM non-essential amino acids and foetal calf serum (FCS) (Life Technologies, Paisley, UK). TRAM-34 was a generous gift from Dr Heike Wulff (University of California Irvine, California, USA).
HLMC purification
HLMC were dispersed enzymatically from macroscopically normal lung obtained within 1 hour of resection for lung cancer and purified using immunoaffinity magnetic selection (Dynabeads) as described previously.10 The final mast cell purity, assessed using metachromatic staining, was >99% with cell viability >98% (monitored by exclusion of trypan blue). HLMC were cultured in DMEM/Glutamax/Hepes containing 1% antibiotic/antimycotic solution, 1% non-essential amino acids, 10% FCS, 100 ng/ml SCF, 50 ng/ml IL-6, and 10 ng/ml IL-10.
HLMC proliferation assay
Following purification, HLMC were resuspended in DMEM/Glutamax/Hepes containing SCF (100 ng/ml), interleukin (IL)-6 (50 ng/ml), and IL-10 (10 ng/ml) at a concentration of 0.25×106 cells/ml as described previously.16 Clotrimazole and TRAM-34 were added in the concentration range 10–1000 nM. Control wells containing either 0.1% dimethyl sulphoxide (DMSO) or culture medium alone were also established. Metachromatic cells were counted after 1 and 4 weeks in culture using Kimura stain.
HLMC chemotaxis
HLMC chemotaxis assays were performed using the Transwell system (BD Biosciences, Oxford, UK) with 24 well plates as described previously.4,5 CXCL10 or SCF was placed in the lower wells (omitted in negative control) at a concentration of 100 ng/ml. 50 μl of 2× the final concentration of TRAM-34 (final concentration 20 or 200 nM), charybdotoxin or iberiotoxin (final concentration of 100 nM) was added to the upper chambers immediately before the addition of 1×105 HLMC per well (50 μl). The vehicle for TRAM-34 was DMSO and the final concentration of DMSO was 0.1% in all wells including a control. After incubating the cells for 3 hours at 37°C, the number of HLMC in the bottom well was counted using Kimura stain in a haemocytometer. HLMC migration was calculated as the fold increase of migrated cells in the test wells compared with the negative control (no chemoattractant in the lower well) as described previously.4,5
ASM cells (passage 3–7) from subjects with asthma (n = 4) were plated into six-well plates (9.6×104 cells/2 ml DMEM, 10% fetal calf serum), grown for 1 week, then growth was arrested for 48 hours with serum deprived medium and stimulated with tumour necrosis factor (TNF)-α (10 ng/ml) for 24 hours.5 The supernatants were removed and stored at −80°C before use in the chemotaxis experiments.
Patch clamp electrophysiology
The whole cell variant of the patch clamp technique was used.10 Patch pipettes were made from borosilicate fibre containing glass (Clark Electromedical Instruments, Reading, UK) and their tips were heat polished resulting in resistances of typically 4–6 MΩ. The standard pipette solution contained (in mM): KCl, 140; MgCl2, 2; HEPES, 10; Na+ATP, 2; GTP, 0.1; pH 7.3. The standard external solution contained (in mM): NaCl, 140; KCl, 5, CaCl2, 2; MgCl2,1; HEPES, 10; pH 7.3. For recording, mast cells were placed in 35 mm dishes containing standard external solution.
Whole cell currents were recorded using an Axoclamp 200A amplifier (Axon Instruments, Foster City, CA, USA), and currents evoked by applying voltage commands to a range of potentials (120 to +130 mV) in 10 mV steps from a holding potential of −20 mV. The currents were digitised (sampled at a frequency of 10 kHz), stored on computer, and subsequently analysed using pClamp software (Axon Instruments). Capacitance transients were minimised using the capacitance neutralisation circuits on the amplifier. Correction for series resistance was not routinely applied. In some experiments continuous membrane currents were recorded at a constant holding potential of +40 mV, data being digitised at 200 Hz and recorded using Axoscope (Axon Instruments). Experiments were performed at 27°C, the temperature being controlled by a Peltier device. Experiments were performed with a perfusion system (Automate Scientific Inc, San Francisco, CA, USA) to allow solution changes, although drugs were added directly to the recording chamber.
Data presentation and statistical analysis
Data are presented as mean (SE) values from separate donors performed in duplicate. Confidence intervals were calculated at 95% using Graphpad Prism 4 software. The inhibition of chemotaxis is presented as the percentage migration compared with the positive control after the subtraction of the negative control from all conditions. Differences between data sets were analysed using a paired two tailed Student’s t test (Microsoft Excel v2003). p values of <0.05 were considered statistically significant.
All human subjects gave written informed consent and the study was approved by the Leicestershire Research Ethics Committee.
RESULTS
HLMC proliferation
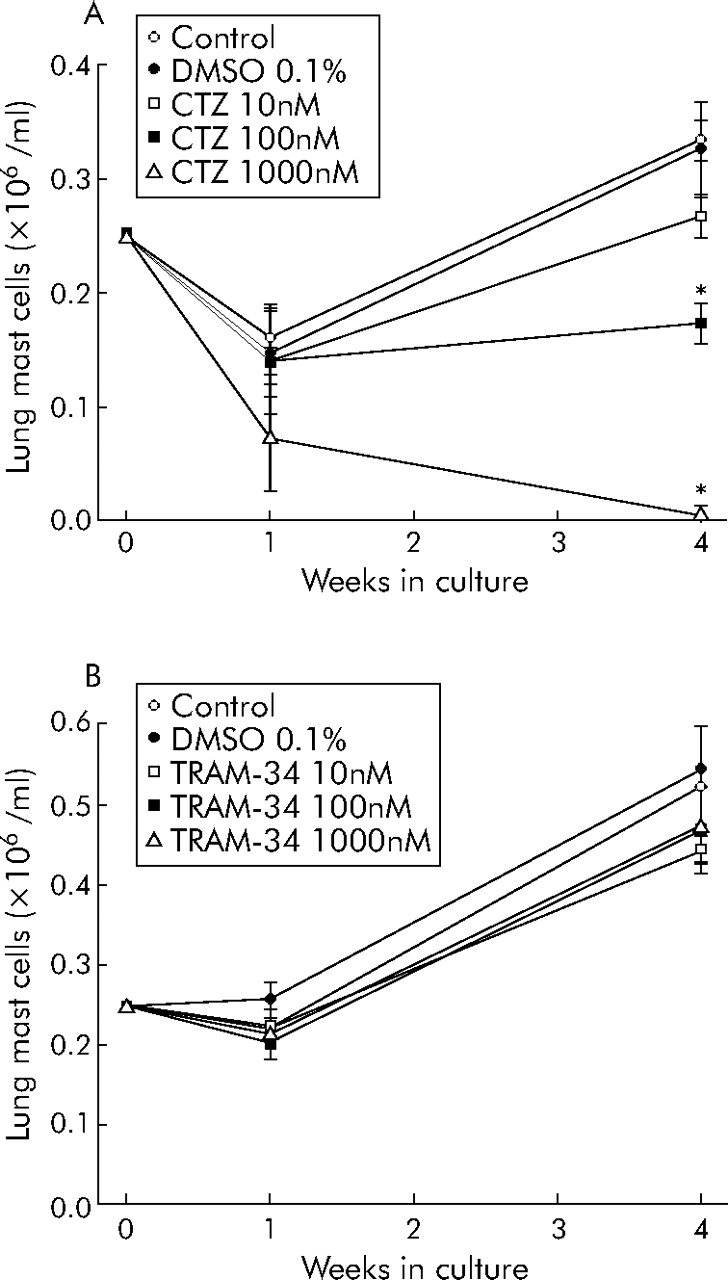
HLMC proliferate in long term culture.16 Clotrimazole, a blocker of KCa3.1 (Kd 70 nM) and inhibitor of cytochrome P450, produced a dose dependent reduction in HLMC number after 4 weeks in culture, with complete cell death evident at 1000 nM (fig 1A⇓). In contrast, TRAM-34, a highly specific blocker of KCa3.1 (Kd 20 nM) without any effect on cytochrome P450, had no effect on HLMC at 1 or 4 weeks (fig 1B⇓). This suggests that KCa3.1 is not involved in HLMC proliferation and that the effects of clotrimazole were “toxic” and independent of its effect on KCa3.1. This is in contrast to the role of KCa3.1 in human T lymphocyte proliferation.15
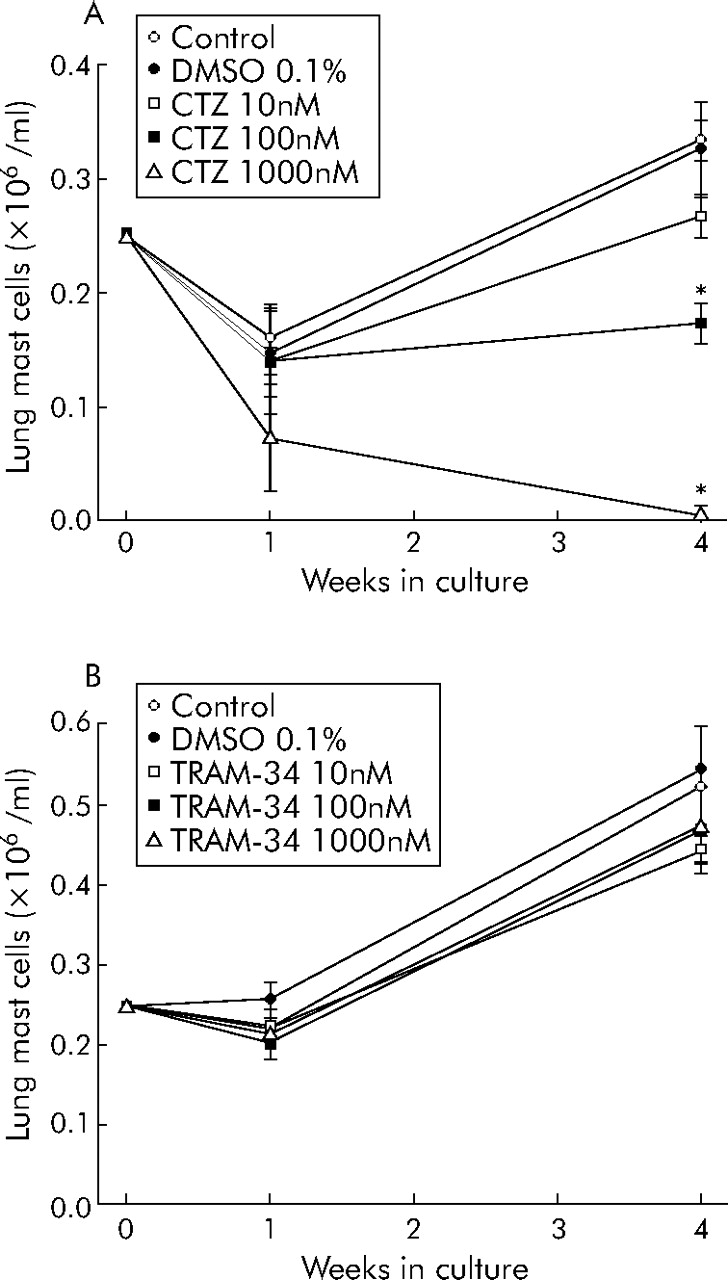
HLMC proliferation in long term culture. (A) HLMC proliferation was attenuated dose dependently by the addition of clotrimazole (CTZ). Higher concentrations (μM) of clotrimazole killed the cells. (B) Conversely, triarylmethane-34 (TRAM-34) had no significant effect on HLMC proliferation. Drug concentrations are given in nmol/l and data are mean (SE) values from three separate donors. Donors in (A) are different from those in (B). *p<0.05.
HLMC chemotaxis
Because of its apparent toxic effect on HLMC, clotrimazole was not used in the chemotaxis assays. For blockade of KCa3.1 during chemotaxis we used TRAM-34 and another KCa3.1 blocker, charybdotoxin (Kd 5 nM).
The migration of HLMC in response to 100 ng/ml CXCL10 was 2.2 (0.2) fold greater than that of the control (no CXCL10, n = 6; 95% CI 1.7 to 2.7, p = 0.001, fig 2⇓), in keeping with previous experiments.4 CXCL10 induced migration was inhibited dose dependently by TRAM-34. Thus, with 200 nM TRAM-34 migration was reduced by 80 (7)% (n = 6; 95% CI 62 to 99, p = 0.0001, fig 3⇓). Consistent with this, CXCL10 induced HLMC migration was inhibited by 78 (14)% with the addition of 100 nM charybdotoxin (n = 6; 95% CI 42 to 113, p = 0.002, fig 3⇓).
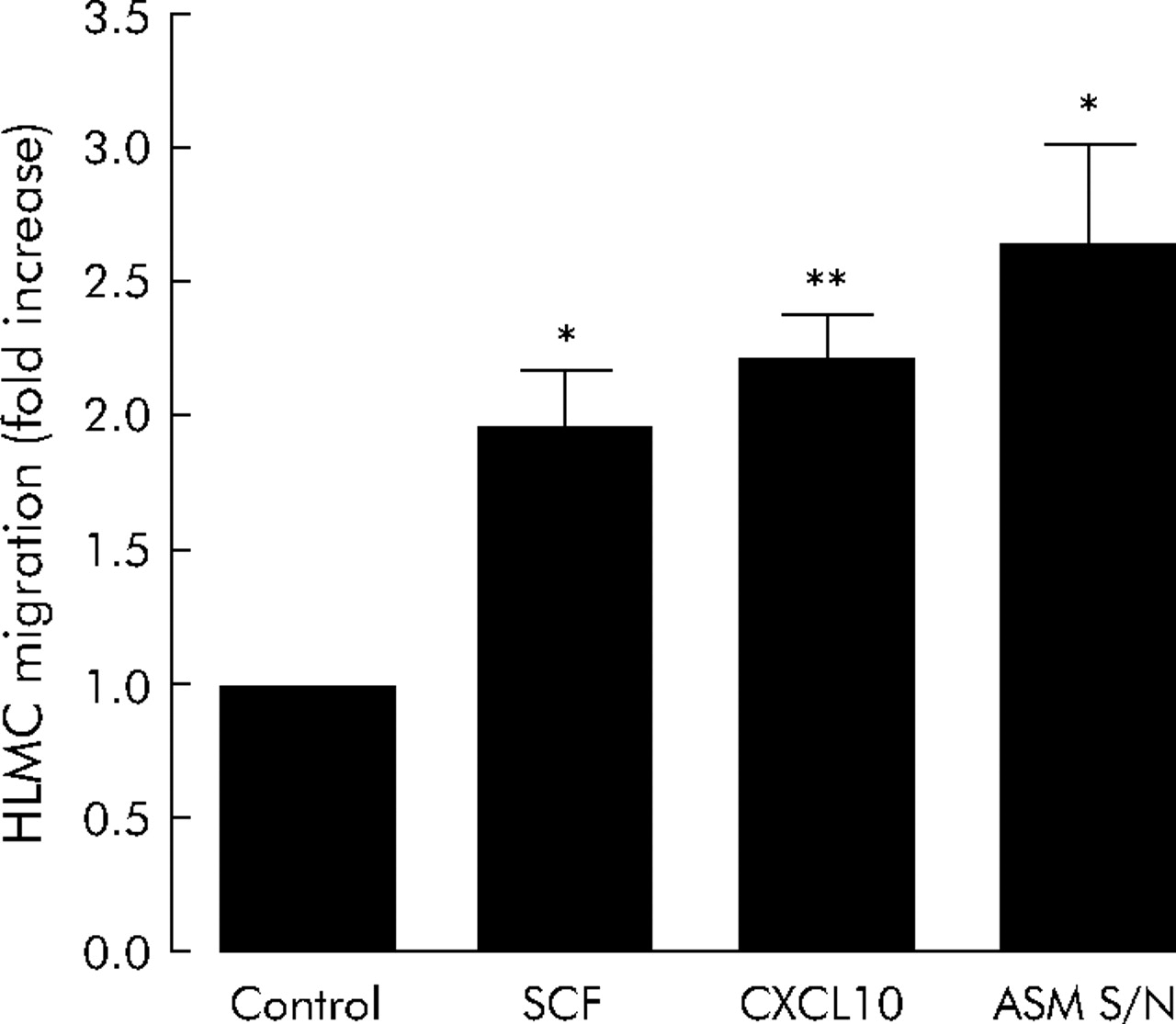
Effect of stem cell factor (SCF, n = 4), CXCL10 (n = 6), and supernatants from tumour necrosis factor (TNF) α stimulated asthmatic airway smooth muscle (ASM, n = 4) on HLMC migration. Data are presented as mean (SE) values from at least four individual donors. *p<0.05; **p<0.01.

Attenuation of CXCL10 induced HLMC migration with KCa3.1 blockade. Triarylmethane-34 (TRAM-34) attenuated CXCL-10 induced HLMC migration in a dose dependent manner. The pharmacologically distinct KCa3.1 blocker charybdotoxin (ChTX) attenuated CXCL10 induced HLMC migration with similar efficacy to TRAM-34. Data are mean (SE) values from six separate donors. *p<0.005; **p<0.001.
To determine whether the inhibition of HLMC migration by KCa3.1 blockade was restricted to an interaction with G protein coupled chemoattractants or a general phenomenon, we next tested the effects of TRAM-34 and charybdotoxin on SCF induced migration. SCF (100 ng/ml) induced HLMC migration to a similar extent as CXCL10 in the majority of donors with mean migration of 2.0 (0.2) fold compared with control (n = 4, 95% CI 1.3 to 2.6, p = 0.020, fig 2⇑). Charybdotoxin (100 nM) and TRAM-34 (200 nM) inhibited SCF induced migration by 92 (8)% (n = 4, 95% CI 69 to 116, p = 0.001), and 76 (11)% (n = 4, 95% CI 42 to 110, p = 0.006), respectively (fig 4⇓).

Attenuation of stem cell factor (SCF) induced HLMC migration with KCa3.1 blockade. Blockade of KCa3.1 attenuated SCF induced HLMC migration with similar efficacy to CXCL10 induced HLMC migration. Data are mean (SE) from four separate donors. TRAM-34, triarylmethane-34; ChTX, charybdotoxin. *p<0.05; **p<0.01.
As a further control we also examined the effects of the specific large conductance Ca2+ activated K+ channel (KCa1.1, also known as BKCa) blocker iberiotoxin (100 nM). Since HLMC do not express KCa1.1 currents or mRNA,10,22 iberiotoxin should be ineffective. Indeed, iberiotoxin did not attenuate the migration of HLMC to either CXCL10 or SCF (2.0 (0.6) fold in the control compared with 1.8 (0.5) fold with 100 nM iberiotoxin). Background control migration was not inhibited by charybdotoxin or TRAM-34.
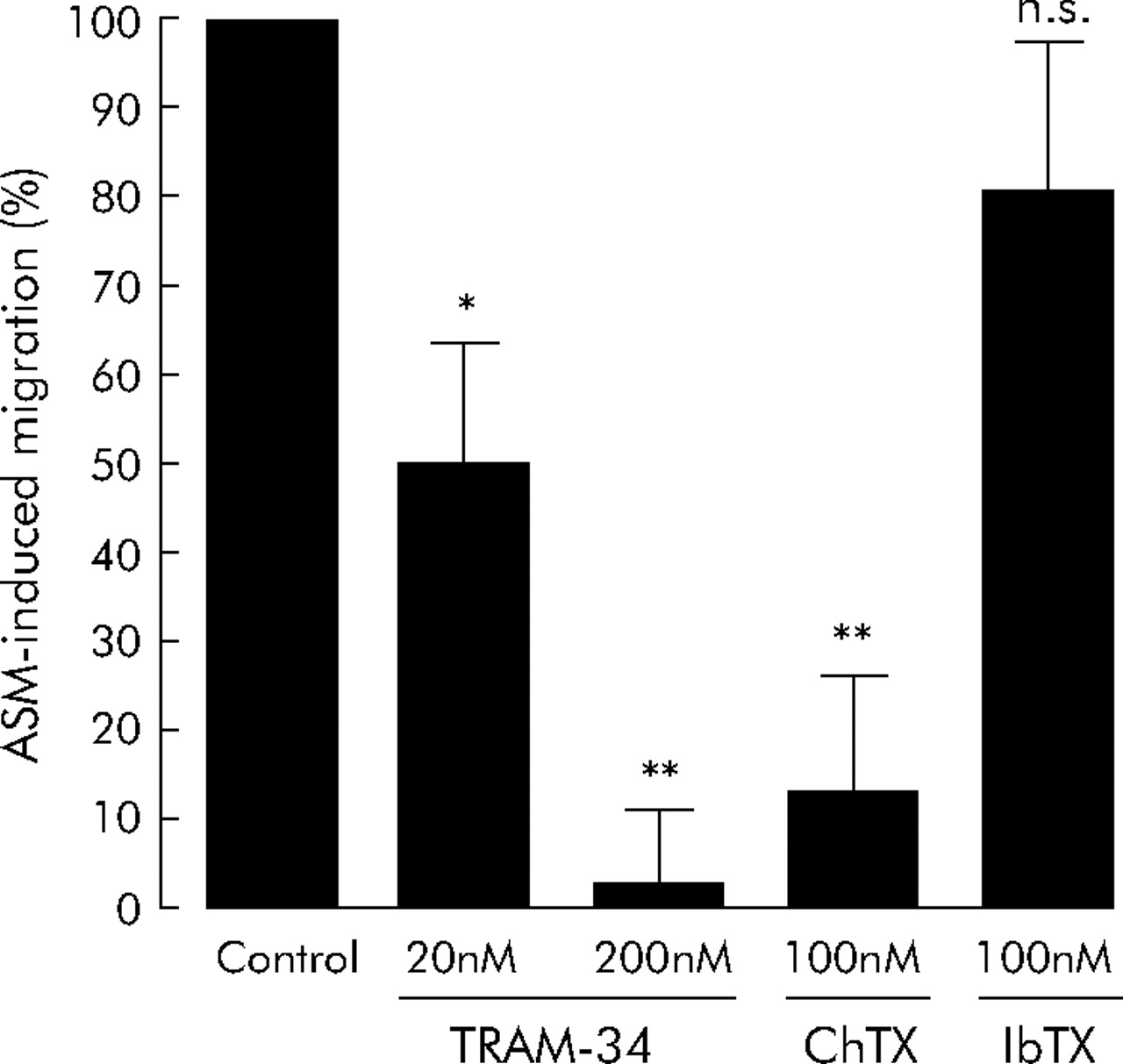
Since HLMC migration to SCF and CXCL10 was inhibited with similar efficacy, we next examined the effects of KCa3.1 blockade on HLMC migration towards the complex milieu of chemoattractants present in cell supernatants from stimulated asthmatic ASM. The ASM supernatants induced HLMC migration to a greater extent than either CXCL10 or SCF alone, with mean migration of 2.6 (0.4) fold compared with control (n = 4, 95% CI 1.5 to 3.8, p = 0.022, fig 2⇑). Both charybdotoxin (100 nM) and TRAM-34 (200 nM) inhibited HLMC migration to ASM supernatants with similar efficacy as with CXCL10 or SCF alone, with 87 (12)% inhibition of migration with charybdotoxin (n = 4, 95% CI 47 to 126, p = 0.006) and 97 (8)% inhibition with TRAM-34 (n = 4, 95% CI 72 to 123, p = 0.001, fig 5⇓). In contrast, iberiotoxin did not attenuate the migration of HLMC towards ASM supernatants (2.6 (0.4) fold in the control compared with 2.4 (0.5) fold with 100 nM iberiotoxin) (n = 4, 95% CI 0.9 to 4.0, p = 0.538, fig 5⇓).

Attenuation of HLMC migration towards tumour necrosis factor (TNF)-α stimulated asthmatic ASM supernatants with KCa3.1 blockade. Triarylmethane-34 (TRAM-34) attenuated HLMC migration to ASM supernatants in a dose dependent manner. Charybdotoxin (ChTX) inhibited migration with similar efficacy to TRAM-34. Iberiotoxin (IbTX), which is structurally related to charybdotoxin but does not block KCa3.1, had no significant effect on HLMC migration. Data are presented as mean (SE) values from four separate donors. *p<0.05; **p<0.01; NS, not significant.
Effects of CXCL10 on HLMC KCa3.1 activation
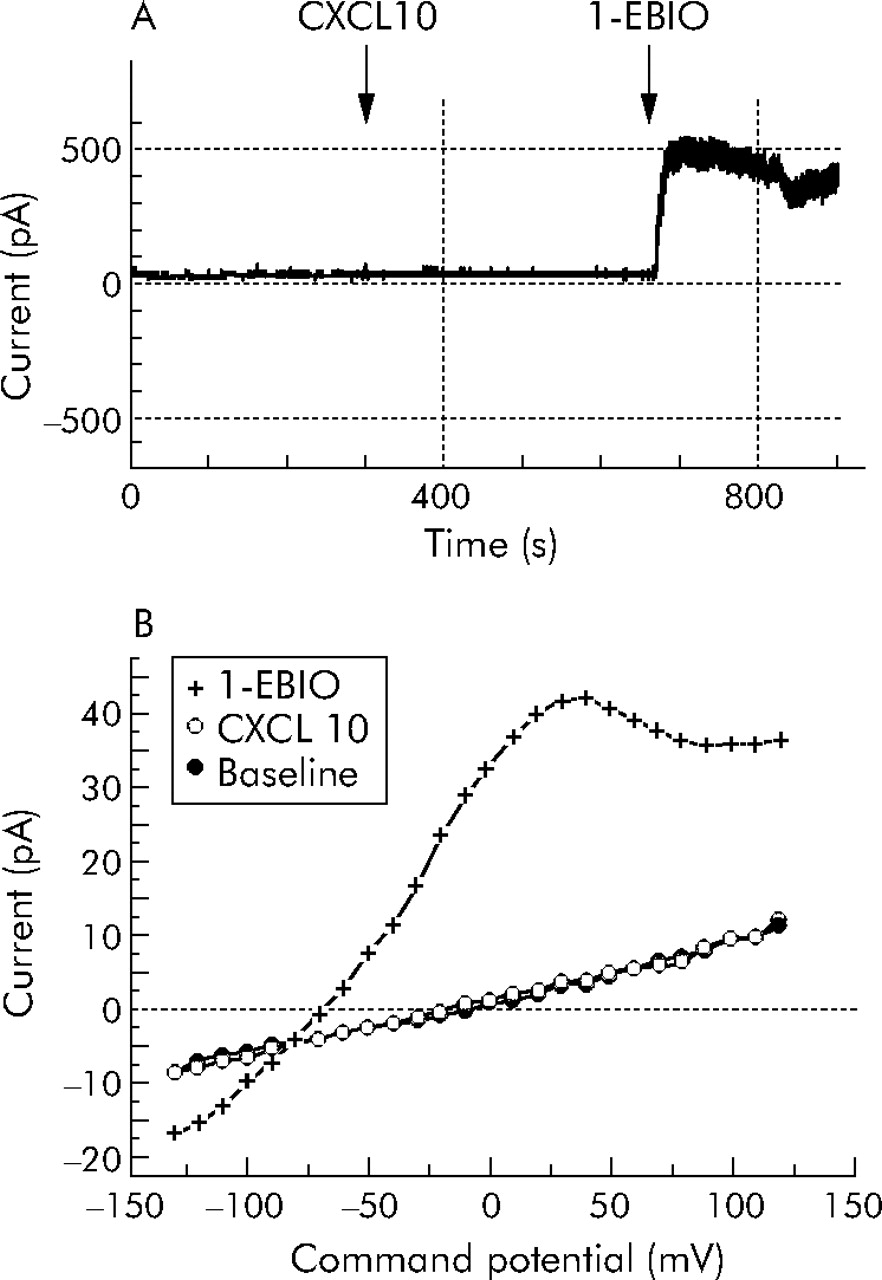
We have recently shown that the Gαs coupled β2 adrenoceptor influences KCa3.1 gating.17 To assess whether the Gαi coupled CXCL10 receptor CXCR3 influences KCa3.1 function we attempted to record KCa3.1 currents following the addition of 100 ng/ml CXCL10 to HLMC (n = 23 cells from four separate donors). In spite of being able to record KCa3.1 currents in these cells following the addition of the KCa3.1 specific channel opener 1-ethyl-2-benzimidazolinone (1-EBIO), we did not see any evidence of either the transient or sustained opening of KCa3.1 in response to CXCL10 (fig 6⇓).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Whole cell electrophysiological recording of electrical currents in HLMC at rest and following the addition of CXCL10 and then 1-ethyl-2-benzimidazolinone (1-EBIO). (A) Continuous recording of current at +40 mV in an HLMC showing a stable baseline with minimal basal current. No response was seen to the addition of CXCL10 where indicated, but a rapid increase in outward current occurred within 90 seconds of adding 1-EBIO to the recording chamber. (B) Current-voltage curves from another cell at rest 3 minutes after the addition of CXCL10 and then 3 minutes after the addition of 1-EBIO showing development of a typical KCa3.1 whole cell current following the latter.
DISCUSSION
In this study we have shown for the first time that blockade of the K+ channel KCa3.1 markedly attenuates the chemotactic response of ex vivo HLMC to the chemokine CXCL10, the mast cell growth factor SCF, and conditioned media from asthmatic ASM. In contrast to its effects on acute mitogen induced lymphocyte proliferation, KCa3.1 does not appear to be important for HLMC proliferation in long term culture.
The redistribution of mast cells within human tissues is likely to be key in the initiation and propagation of a variety of diseases.2,3,5 Inhibiting their migration within tissues may therefore offer a truly novel approach for the treatment of mast cell driven pathobiology. As an example, we have recently shown that the CXCL10/CXCR3 axis may be a critical determinant of mast cell distribution within the human lung.5 Thus, inhibition of CXCL10/CXCR3 induced chemotaxis is an attractive target for asthma therapy. However, while targeting CXCL10/CXCR3 may prove fruitful, the ASM produces several other chemoattractants including SCF, TGFβ and other chemokines.5–,7 These may play their own part in mast cell recruitment by ASM under certain conditions and, in addition, are probably important for the microlocalisation of mast cells within other tissue compartments in unrelated diseases such as tubulointerstitial renal disease and rheumatoid arthritis.2,3,18 The ability of KCa3.1 blockade profoundly to inhibit mast cell migration in response to two diverse chemoattractants—CXCL10 which activates the G protein coupled receptor CXCR3 and SCF which activates the tyrosine kinase receptor CD117—therefore suggests that KCa3.1 has great potential as a therapeutic target for mast cell mediated disease in humans. Moreover, blockade of KCa3.1 almost completely attenuates HLMC migration towards the complex milieu of chemoattractants present in TNFα stimulated asthmatic ASM cell supernatants, strengthening the hypothesis that functional KCa3.1 channels are an absolute requirement for HLMC migration.
To inhibit HLMC migration we used charybdotoxin and TRAM-34, two distinct molecules which block the KCa3.1 pore at different sites.19,20 Charybdotoxin is a 37 amino acid peptide derived from the venom of the scorpion Leiurus quinquestriatus and blocks KCa3.1 with a Kd for channel block of 5–10 nM.19,21 It also blocks the voltage gated K+ channel Kv1.3 and the large conductance KCa, KCa1.1. However, HLMC do not express Kv1.3 or KCa1.1 mRNA or their electrical currents.10,22 TRAM-34 is a highly specific small molecule blocker of KCa3.1 (Kd 20 nM)23 which was derived from the KCa3.1 blocker and antifungal agent clotrimazole. However, unlike clotrimazole, TRAM-34 does not interfere with cytochrome P450.23 Charybdotoxin blocks KCa3.1 by binding to the external pore with high affinity,19 while the highly lipophilic TRAM-34 binds to residues within the internal vestibule of the channel.20 We used TRAM-34 and charybdotoxin at concentrations up to 10 times the Kd because it has been estimated previously that, for complete channel block to be achieved, drugs need to be present at 5–10 times the Kd.15 The ability of these two potent but pharmacologically distinct KCa3.1 blockers to inhibit HLMC migration to a similar extent at 10 times the Kd therefore indicates that the mechanism behind this is indeed KCa3.1 blockade. In addition, the KCa1.1 blocker iberiotoxin was without effect, further suggesting that the effects observed are specific to KCa3.1.
The role of KCa3.1 in HLMC and T cell mediated secretion is to maintain the negative membrane potential during cell activation, counteracting the tendency for Ca2+ influx to depolarise the cell membrane. Thus, KCa3.1 increases the driving force for Ca2+ influx because store operated Ca2+ channels conduct larger currents at negative membrane potentials.10,11,13 However, the role of this channel in cell migration is predicted to be different. A KCa channel with some properties of KCa3.1 has been demonstrated in glial cells and has been proposed to facilitate the retraction of the rear body of the migrating cell,24,25 despite appearing to be more highly expressed at the leading edge of lamellipodia in migrating MDCK-F (transformed renal epithelial cells), NIH-3T3 fibroblasts, and human melanoma cells.26 The mechanism controlling their seemingly selective activation at the rear body of the cell is unclear, although the localised Ca2+ concentration is likely to be critical. In addition, migration appears to require the opening and closing of the channels, most probably due to calcium oscillations, since both KCa3.1 channel blockers and the opener 1-EBIO all inhibit chemotaxis in these cells.14,24 The intermittent activation of the KCa3.1-like channel in these cells is therefore believed to facilitate the swelling and shrinking of the cell body required for cell migration,25 and would explain why functional KCa3.1 are important for migration in HLMC.
CXCL10 increases intracellular Ca2+ transiently in HLMC through the release of Ca2+ from internal stores but does not activate Ca2+ influx from the extracellular fluid.4 We have shown in this study that CXCL10 does not directly open KCa3.1, which is in keeping with our previous finding that influx of extracellular Ca2+ is the critical requirement for KCa3.1 opening during HLMC activation.10 In addition, SCF does not open KCa3.1.10 This indicates that the gating of KCa3.1 during HLMC migration is downstream of the chemoattractant stimulus, most probably related to adhesive signals required for the migratory process. These data also show that, while the KCa3.1 channel in HLMC is known to be directly coupled to the Gαs dependent β2 adrenoceptor,17 it is not coupled to the Gαi dependent CXCR3 receptor.
We have previously shown that HLMC proliferate in long term culture. Following an initial decrease in cell number over the first week, HLMC proliferate so that by 4 weeks there can be up to four times the starting number.16 Interestingly, in contrast to published observations in T and B cells27,28 and endothelial cells,29 TRAM-34 had no significant effect on HLMC proliferation. TRAM-34 inhibits mitogenesis of preactivated human T cells at concentrations similar to those required for channel blocking.27 In contrast to TRAM-34, clotrimazole did inhibit proliferation and, at a concentration of 1000 nM, killed HLMC, suggesting that this effect was independent of KCa3.1 blockade (fig 1A⇑). Clotrimazole has many diverse effects on cells other than channel blocking—for example, reducing the expression of G1 phase cyclins,30 modulation of cytochrome P450 activity,31 and inhibition of cellular glycolysis32—all of which could contribute to the antiproliferative effects presented here. These data suggest that SCF induced HLMC proliferation does not involve KCa3.1, unlike acute T cell mitogenic stimulation with anti-CD3.15
Blockade of KCa3.1 has shown promise in several diseases/disease models. Clotrimazole was very effective in ameliorating active human rheumatoid arthritis but caused unacceptable side effects due to inhibition of cytochrome P450.33 TRAM-34 prevents vascular restenosis after balloon angioplasty in rats through its ability to inhibit neointimal vascular smooth muscle proliferation and without any undue toxicity.34 Another specific KCa3.1 blocker, 4-phenyl-4H-pyran (Kd 8 nM), reduces infarct volume in a rat model of subdural haematoma, suggesting a possible use in the management of traumatic brain injury.35 Clinical trials are also underway to study the effects of KCa3.1 inhibition in sickle cell anaemia. These studies of KCa3.1 blockade are encouraging, and the lack of obvious toxicity of TRAM-34 suggests real therapeutic potential for human disease. With regard to mast cell mediated disease, however, further mechanistic studies of KCa3.1 blockade using rodent models are unlikely to be informative because KCa3.1 currents have not been observed in rodent mast cells.8
In summary, we have shown for the first time that blockade of the K+ channel KCa3.1 markedly attenuates the chemotactic response of ex vivo HLMC to both the chemokine CXCL10 and the mast cell growth factor SCF, as well as TNFα stimulated asthmatic ASM supernatants. This suggests that blocking KCa3.1 has great potential as a target for the treatment of asthma and other inflammatory diseases in which mast cells play a role.
REFERENCES
Footnotes
Published Online First 29 June 2006
This study was supported by the Wellcome Trust.
Competing interests: none.