Article Text

Abstract
Cigarette smoke causes significant oxidant stress which is further enhanced by recruitment and activation of inflammatory cells to the lung. Polymorphisms in some detoxification enzymes are thought to increase the risk of developing chronic obstructive pulmonary disease (COPD), but the ultimate role of genetic variability in antioxidant and/or detoxification enzymes in COPD remains obscure. Some antioxidant enzymes are inducted, but the extent of induction is insufficient to protect the lung/alveolar epithelium against cigarette smoke. Exogenous antioxidants such as vitamins do not seem to protect against cigarette smoke related lung injury. Glutathione related synthetic drugs such as N-acetylcysteine have shown some benefits, but they may have pro-oxidant side effects. Synthetic compounds with superoxide dismutase and catalase activities have shown promising results in animal models against a variety of oxidant exposures including cigarette smoke in the lung. These results are in agreement with studies highlighting the importance of alveolar antioxidant protection mechanisms in oxidant stress and their inducibility. These new drugs need to be tested in cigarette smoking related lung injury/inflammation since inflammation/oxidant stress can continue after discontinuation of smoking.
- COPD, chronic obstructive pulmonary disease
- CuZnSOD, copper zinc SOD, ECSOD, extracellular SOD
- ELF, epithelial lining fluid
- GCL, glutamate cysteine ligase
- GPX, glutathione peroxidase
- GR, glutathione reductase
- GRX, glutaredoxin
- GSH, glutathione
- GST, glutathione-S-transferase
- γGT, γ-glutamyl transpeptidase
- HO-1, heme oxygenase-1
- MnSOD, manganese SOD
- MRP, multidrug resistance protein
- NAC, N-acetylcysteine
- NO, nitric oxide
- PRX, peroxiredoxin
- ROS, reactive oxygen species
- RNS, reactive nitrogen species
- SOD, superoxide dismutase
- TRX, thioredoxin
- chronic obstructive pulmonary disease
- antioxidant enzymes
- smoking
- smoker’s lung
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- CuZnSOD, copper zinc SOD, ECSOD, extracellular SOD
- ELF, epithelial lining fluid
- GCL, glutamate cysteine ligase
- GPX, glutathione peroxidase
- GR, glutathione reductase
- GRX, glutaredoxin
- GSH, glutathione
- GST, glutathione-S-transferase
- γGT, γ-glutamyl transpeptidase
- HO-1, heme oxygenase-1
- MnSOD, manganese SOD
- MRP, multidrug resistance protein
- NAC, N-acetylcysteine
- NO, nitric oxide
- PRX, peroxiredoxin
- ROS, reactive oxygen species
- RNS, reactive nitrogen species
- SOD, superoxide dismutase
- TRX, thioredoxin
The most important non-malignant lung disease caused by cigarette smoke is chronic obstructive lung disease (COPD), a globally escalating problem that mirrors the extent of cigarette smoking in the community. In addition to COPD, cigarette smoking is associated with respiratory bronchiolitis interstitial pneumonia (RB-ILD)/desquamative interstitial pneumonia (DIP), a disease entity classified as a histopathological group of interstitial pneumonias.1 Both disease groups are underdiagnosed; this is a serious problem, especially for COPD because of its high prevalence, progressive nature, and cost to healthcare systems throughout the world. Smoking cessation is the only therapeutic intervention that has been shown to improve the prognosis of COPD with reduction of disease progression. This review will concentrate on the importance of the oxidant/antioxidant balance and the major antioxidant enzymes and detoxification mechanisms in smoker’s lung and COPD, and the possibilities that new synthetic antioxidants will be able to prevent the progression of cigarette smoke mediated injury in the human lung.
The lung is a unique tissue compared with many other vital organs since it is directly exposed to high levels of oxygen. It therefore has to have efficient antioxidant mechanisms for the primary protection of the airways against ambient oxygen and exogenous oxidants. In cigarette smokers the oxidant burden is further increased for a number of reasons. Firstly, one puff of cigarette smoke contains millions of oxygen free radicals. Cigarette smoke contains high concentrations of nitric oxide (NO). Tar includes organic compounds such as quinones, which react with oxygen and increase the generation of superoxide radicals in intracellular redox cycling reactions. Free radicals activate inflammatory cells which, in turn, generate high levels of reactive oxygen (ROS) and nitrogen species (RNS) and other toxic metabolites. The most important of these mechanisms include myeloperoxidase, NADPH oxidase, and nitric oxide synthases (NOSs) (such as inducible NOS) and, in certain inflammatory conditions, eosinophil peroxidase. Typical features of smoker’s lung are inflammation with the recruitment of neutrophils and macrophages into the airways, small airway obstruction/fibrosis, and destructive processes of the lung parenchyma.2,3 Some studies have suggested that oxidative stress/lipid peroxidation is associated with pulmonary airway narrowing in the general population.4,5 Many studies have also shown that there is a significant oxidant burden both in the lung and systemic circulation in COPD.6–11 The airways of patients with COPD, especially during disease exacerbations, contain high levels of activated neutrophils, activated macrophages, and also eosinophils, representing an even higher oxidant burden during the active phase of this disease.3,12 A number of studies have detected increased levels of oxidant markers in COPD lung tissue as well as in sputum, exhaled air, and exhaled breath condensate. These markers include increased levels of myeloperoxidase, the neutrophil markers lipocalin and lactoferrin, and markers of lipid peroxidation such as 4-hydroxy-2-nonenal (4-HNE) in the tissue and sputum, increased levels of hydrogen peroxide, and markers of lipid peroxidation products (thiobarbituric acid reactive substances) in the exhaled air/breath condensate in COPD. These studies have been extensively reviewed previously.9,13–16 Most studies have been conducted on chronic smokers. However, acute exposure to cigarette smoke has also been shown to lead to an immediate transient oxidant burden, although unchanged and even decreased levels of exhaled NO have also been reported (reviewed by van der Vaart8). Overall, there is a significant ROS/RNS production in COPD that apparently overwhelms lung antioxidant protection in this disease.
In contrast to the oxidant producing systems, relatively little is known about the primary antioxidant defence of human lung, and even less is known about this system in smokers. Furthermore, alterations in the primary antioxidant defence as well as the effects of exogenous antioxidants in COPD have been less extensively investigated. There are many questions that still remain unanswered, such as the importance of individual variability in the primary antioxidant defence and the risk for COPD, the significance of the induction of the primary antioxidant mechanisms in smoke related inflammation, the importance of specific lung cell types in lung protection, the effects of the ultrastructural localisation of these enzyme proteins and/or small molecular weight antioxidants in smoker’s lung, and also whether antioxidant enzymes have any effects on the inflammatory process itself such as the survival of inflammatory cells or apoptosis. The balance between oxidants and antioxidants is complex and the interactions between these pathways have received little attention. However, the significance of the primary antioxidant defence mechanisms in the development of COPD as well as finding ways to improve lung antioxidant protection, especially in areas where the oxidant stress is highest, may be major questions in predicting COPD progression.
ANTIOXIDANT DEFENCE OF HUMAN LUNG
The lung has an efficient protection capacity against exogenous oxygen and ROS. Studies on animal models have shown that mucins, which are cysteine rich glycoproteins present in the epithelial lining fluid, provide significant protection against oxidants, and that the levels of these glycoproteins are increased by oxidants.17,18 Epithelial lining fluid contains a high glutathione (GSH) content,19 a molecule that also has been considered as one of the primary antioxidants in the human lung. In addition to these compounds, lung tissue contains several proteins capable of binding free iron and other metals (albumin, transferrin, ferritin, ceruloplasmin, lactoferrin, metallothionein). These proteins may have fundamental roles in protecting lung tissue against oxidant attack since metals are potent generators of free radicals in the Fenton and superoxide derived Haber-Weiss reactions.20 Human airways contain both lipid and water soluble vitamins, but their relative roles in the airways are poorly understood. One typical feature of the antioxidant enzymes and closely related thiol proteins of human lung is their highly cell specific arrangement and compartmentalisation, in that they are localised to different cell types, cell organelles (one example being mitochondria), and also different extracellular compartments.
Superoxide dismutases
Superoxide dismutases (SODs) are the only enzyme family with activity against superoxide radicals. These enzymes include manganese SOD (MnSOD), copper zinc SOD (CuZnSOD), and extracellular SOD (ECSOD).21–23 All of them are widely expressed in human lung.24 CuZnSOD, which is mainly a cytosolic enzyme, is evenly distributed intracellularly but is also found in the nucleus and lysosomes.25 It is expressed in the bronchial epithelium of human lung,26 and in the alveolar epithelium, mesenchymal cells, fibroblasts and vascular endothelial cells in rat lung.27 MnSOD is localised to the mitochondria, a critical cell organelle both for cellular energy metabolism and cell survival. MnSOD is mainly expressed in alveolar type II epithelial cells and alveolar macrophages in the human lung.26,28 ECSOD is primarily localised to the extracellular matrix, especially in areas containing high amounts of type I collagen fibres and around pulmonary and systemic vessels.29 It has also been detected in the bronchial epithelium, alveolar epithelium, and alveolar macrophages. Overall, CuZnSOD and MnSOD are generally thought to act as bulk scavengers of superoxide radicals. However, the relatively high ECSOD level in the lung with its specific binding to the extracellular matrix components may represent a fundamental component of lung matrix protection. All SODs also have functional polymorphisms with potential consequences to the individual variability for the risk of lung diseases.30 SODs are also induced by cytokines. In this respect, MnSOD is unique since it is highly responsive to multiple extracellular stimuli including cytokines, oxidants and radiation, and this induction is thought to have physiological significance, especially in the protection of alveolar epithelium.24
Destruction of hydrogen peroxide
The major enzymes associated with hydrogen peroxide decomposition include catalase and enzymes associated with GSH metabolism. The latter group of enzymes includes glutathione peroxidases (GPXs), glutathione reductase, glutamate cysteine ligase (GCL)—that is, gammaglutamyl cysteine synthase (γGCS) and glutathione synthase. Catalase, a peroxisomal hydrogen peroxide consuming enzyme, is expressed intracellularly mainly in alveolar macrophages and neutrophils.31,32 Due to the high level of GSH (a tripeptide containing one cysteine amino acid—that is, a thiol group)19,33 in the epithelial lining fluid (ELF), enzymes associated with GSH synthesis are thought to have central roles in lung protection. In contrast to catalase, GPXs, a selenium containing family of enzymes, can break down not only hydrogen peroxide but also lipid peroxides. Five GPX gene products have been characterised: GPX1 is the classsic GPX; GPX2 is gastrointestinal; GPX3 is the plasma GPX that has also been detected in the epithelial lining fluid of human lung; GPX4 is the phospholipid hydroperoxide GPX; and GPX5 is a secreted protein synthesised by the caput epididymis. Of these, GPX3 has been most widely investigated in human lung.34 “Oxidised GSH” (that is, glutathione disulphide (GSSG)) is reduced back to GSH by glutathione reductase; the level of GSSG in normal tissues is very low (below 1%) due to the immediate reaction by glutathione reductase. Another important pathway for GSH replenishment in the lung is, however, its synthesis by GCL and glutathione synthase. The rate limiting enzyme in GSH synthesis, GCL, has been suggested to play a fundamental role in the regulation of lung GSH homeostasis. Mammalian GCL is a heterodimer that contains two subunits—the catalytically active heavy subunit (GCLh) and a light subunit (GCLl) that modulates the affinity of the heavy subunit for substrates and inhibitors. Both subunits are localised to the cytosol and expressed especially in human bronchial epithelium and occasionally in alveolar macrophages.35,36 The regulation of GCLh is still somewhat controversial but it does appear to be induced by oxidants and cytokines and downregulated by transforming growth factor β in vitro.37–39 GCL is cleaved during the apoptotic process of human cells,40–42 but it is not known whether this cleavage also occurs in human lung and lung diseases.
In addition, disposal of hydrogen peroxide is closely associated with several thiol-containing proteins—namely, thioredoxins (TRX1 and TRX2), thioredoxin reductases (TRR), peroxiredoxins (PRX) (which are thioredoxin perixidases), and glutaredoxins (GRX).43–46 All these enzymes are considered to be powerful redox modulators; they also have antioxidant properties, protect cells against high oxygen tension, and participate in cell proliferation and survival. Two TRXs and TRRs have been characterised in human cells, existing in both cytosol and mitochondria. In human lung, TRX and TRR are expressed in bronchial and alveolar epithelium and macrophages.47 Six different PRXs have been found in human cells, differing in their ultrastructural compartmentalisation. Experimental studies have revealed the importance of PRX VI in the protection of alveolar epithelium. Human lung expresses all PRXs in bronchial epithelium, alveolar epithelium, and macrophages.48 PRX V has recently been found to function as a peroxynitrite reductase,49 which means that it may function as a potential protective compound in the development of ROS/RNS mediated lung injury. GRXs (I–II) are also localised in both the cytosol and mitochondria.43 GRX1 has recently been found especially in human alveolar macrophages and, to a lesser degree, in bronchial epithelium. It is downregulated by growth factors and possibly induced transiently by oxidant stress in vitro.50 Thus, there are several redox modulatory thiol containing proteins in human lung that have recently been shown to participate not only in cell protection against oxidants but also in the regulation of cell growth and survival.
Other detoxification pathways
Glutathione-S-transferases (GSTs) and multidrug resistance proteins (MRPs) belong to a group of detoxification enzymes that require intracellular GSH for their catalytic activity. GSTs constitute a protein family with high genetic variability, having potential effects on the development of smoking related non-malignant and malignant diseases. Three mammalian GST families—namely cytosolic, mitochondrial and microsomal—have been characterised.51 The relative importance of GSTs and MRPs in human lung is still unknown. For example, there are nine related MRPs. MRP1, at least, is located in the epithelium at the intersection of many thiol-based protection mechanisms that modulate the extrusion of GSSG from the cells (reviewed by Kruh52). Both these families of detoxification enzymes are often considered to be important in malignant diseases and the resistance of cancer cells to oxidant generating drugs. However, they are also expressed in normal lung, mainly in the airways.53 They protect cells against numerous oxidant generating compounds and drugs, including pollutants, quinones, hydroperoxides, and other end products of oxidative metabolism. MRP substrates include structurally diverse GSH, glucuronate and sulphate conjugates, but the precise mechanism by which GSH participates in MRP mediated efflux of the natural toxic products is currently unknown.52 Gamma-glutamyltranspeptidase (γGT) is a plasma membrane enzyme which is also expressed in lung epithelial cells. The released GSH/GSSG is not freely diffusible and needs first to be decomposed to the amino acids where γGT decomposes the γ-glutamyl bond of GSH. Experimental studies have shown that γGT is induced by oxidant stress,54 and also that γGT deficiency results in increased oxidative stress in normoxia.55 Heme oxygenase-1 is a stress response protein with significant functions in cell protection and homeostasis; this is also an enzyme which can be induced by oxidants and cytokines.56 Thus, in addition to the classical antioxidant enzymes and thiol proteins, human lung contains stress inducible enzymes and detoxification systems which seem to confer primary protection for the human airways. The reactions of the major antioxidant and detoxification enzymes are summarised in fig 1. Overall, there are numerous enzyme systems that participate in the scavenging of ROS in human lung with tight interaction; these enzymes are localised to specific cell types and organelles, mostly in bronchial epithelium, alveolar epithelial type II cells and alveolar macrophages. Some of the enzymes are also bound to the extracellular matrix, providing potential protection to the lung interstitium (fig 2).
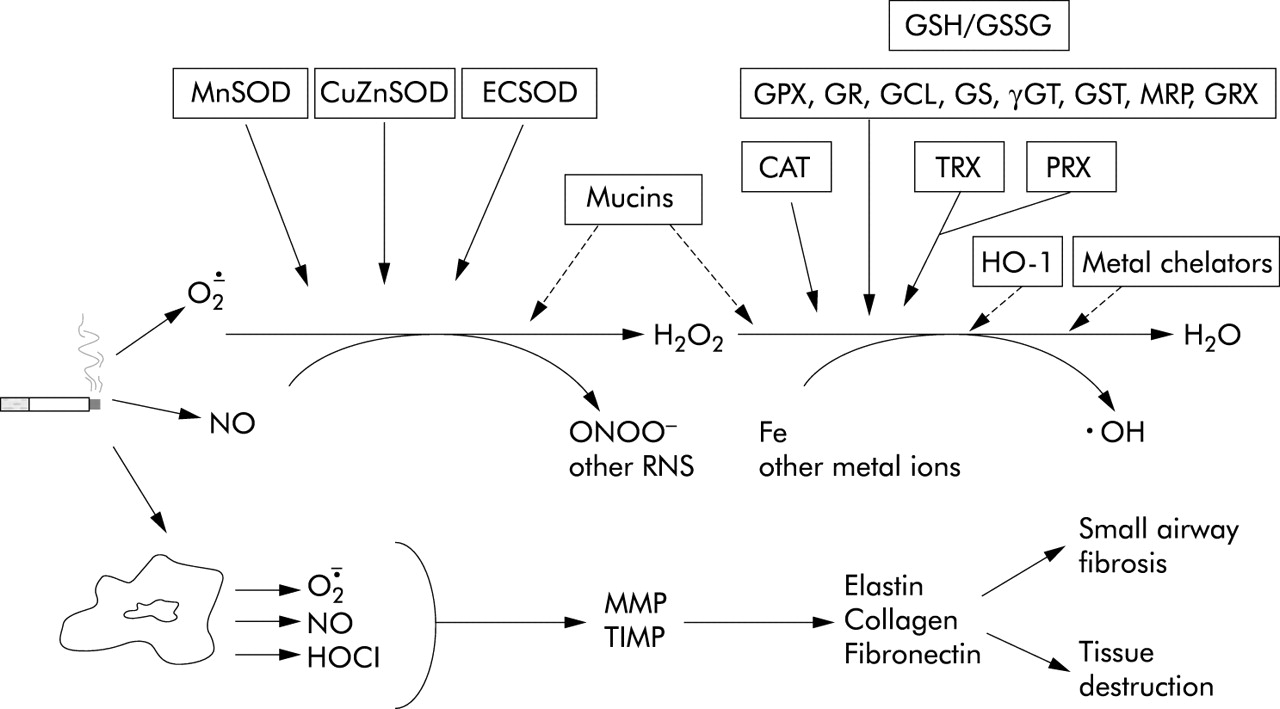
Oxidant generation by cigarette smoke and the major antioxidant defence systems in their disposal. O2·−, superoxide; H2O2, hydrogen peroxide; ·OH, hydroxyl radical; NO, nitric oxide; NOS, nitric oxide synthase; HOCl, hypochlorous acid; ONOO−, peroxynitrite; RNS, reactive nitrogen species; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of matrix metalloproteinase. SOD, superoxide dismutase; CuZnSOD, copper zinc SOD; MnSOD, manganese SOD; ECSOD, extracellular SOD; GPX, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione; GCL, glutamate cysteine ligase; GST, glutathione-S-transferase; GSSG, oxidised glutathione (glutathione disulphide); GS, glutathione synthase; γGT, γ-glutamyltranspeptidase; MRP, multidrug resistance protein; TRX, thioredoxin; PRX, peroxiredoxin; GRX, glutaredoxin; CAT, catalase; HO-1, heme oxygenase-1.

{kind=link}
{kind=link}
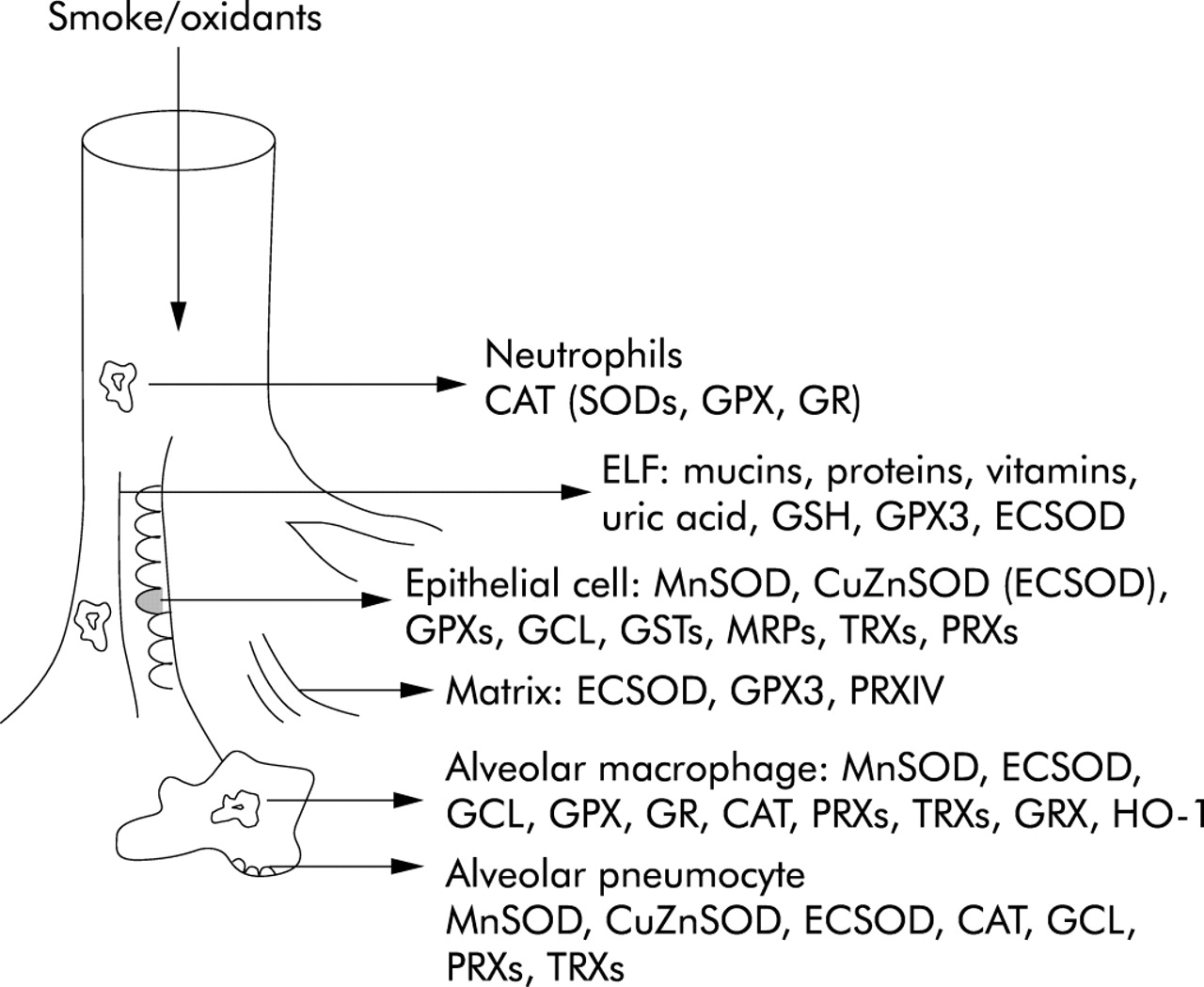
Localisation of major antioxidant defence systems in human lung. ELF, epithelial lining fluid. For abbreviations of antioxidant enzymes see fig 1.
ANTIOXIDANT ENZYMES IN SMOKERS AND SMOKING RELATED LUNG DISEASES
Polymorphisms of antioxidant and detoxification enzymes in COPD
Many antioxidant enzymes such as all three SODs exhibit functional polymorphisms which can impact on enzyme activity/localisation. No studies are available on the polymorphisms of MnSOD or CuZnSOD in the development/progresssion of COPD. Their polymorphisms appear to have no effect on asthma development.57 In ECSOD, a single nucleotide substitution resulting in an amino acid change from Arg to Gly at 213 (in the middle of the positive charged sequence of the heparin binding domain of ECSOD) leads to increased circulating ECSOD activity (10-fold) and decreased tissue binding of ECSOD.58 One preliminary report on this polymorphism suggests that ECSOD polymorphism might be an additional risk for the development of chronic lung disease.59 However, it is likely that polymorphism of ECSOD, which is very rare, cannot alone explain the early development of common lung diseases like COPD. Several studies have suggested that polymorphisms of the GSH associated detoxification enzymes may be associated with COPD. In one study, three GST polymorphisms (GST T1, GST P1 and GST M1) but not hemeoxygenase-1 showed a significant association with the rapid decline in lung function parameters in smokers.60 In some, but not all, studies there were also significant associations between GST M1 null genotypes, GST P1, microsomal epoxide hydrolase, and COPD.61–63 Overall, it appears that GST genotypes are risk factors for a rapid decline or low function in smokers with mild to moderate airflow obstruction.64
Regulation of antioxidant enzymes in smoker’s lung, COPD, and smoking related desquamative interstitial pneumonia
A recent microarray study on the bronchial brushings assessed 44 antioxidant related genes in four categories (catalase, SOD/GSH metabolism/redox balance, and pentose phosphate cycle) in non-smokers and smokers (average smoking history 21 years, no lung disease).65 There was some variation in the gene expression levels within the same individual, but a remarkable interindividual variability. Altogether, 16 of the 44 antioxidant related genes were increased in the epithelium of smokers compared with non-smokers.65 Neither SODs nor catalase were altered in the epithelium of smokers, while 33% (7 of 21) of the genes encoding enzymes of the GSH pathway were upregulated, the most prominent being GPX2, although others with minor induction included GCL (both subunits), glutathione reductase, GPX3, GSTs and—among the redox balance regulating enzymes—TRR 1. Another recent study that used microarray technology characterised the gene expression profile of lung tissue samples from healthy subjects and advanced emphysema.66 A typical feature of severe emphysema, both in “usual emphysema”and alpha-1-antitrypsin deficiency, was a global decrease in gene expression but an increased expression of the transcipts encoding proteins involved in inflammation and proteolysis. The major finding among the classical antioxidant enzymes was a significant decline in GPX3. Overall, GSH associated enzymes represent a group of enzymes that can be induced by cigarette smoke in human lung but may probably also decline during the progression of COPD.
MnSOD is significantly induced by cigarette smoke in rat lungs,67 which is consistent with the high inducibility of MnSOD by cytokines and oxidant exposure in vivo.24 Many studies have, however, also observed that the induction is transient, and that antioxidant enzymes may also be downregulated during severe and/or chronic oxidant exposure.68 Superoxide dismutase and catalase activities have been reported to be increased in human alveolar macrophages in chronic smokers,69 whereas several antioxidant enzymes such as CuZnSOD, GST, and GPX seem to decline in elderly smokers with long smoking histories.70 A recent human study evaluated MnSOD in smokers and in subjects with mild/moderate COPD and found MnSOD to be increased especially in the alveolar pneumocyte II cells of smokers and patients with COPD.71 In conclusion, it seems that MnSOD can be induced in smoker’s lung, especially in the alveolar epithelium, and that this is probably a consequence of cytokine/oxidant mediated induction. Furthermore, it can be viewed as one indicator of the oxidant burden, especially at the alveolar level of smokers.
Cigarette smoke causes depletion of intracellular GSH in cultured airway epithelial cells with a transient decrease of GPX and glucose-6-phosphatase activities,72 but also an increase in GPX 3 expression.73 Some in vitro studies also suggest that, after initial GSH depletion, there is an increase in the GSH levels apparently due to GCL induction (reviewed by Rahman and MacNee74). In general, the results obtained from various animal species and cell cultures are, however, variable, which emphasises the importance of not generalising results from in vitro or animal experiments to human disease states. In humans, these enzymes have been investigated both after acute exposure to smoke and in chronic smokers. Acute smoking, which evidently causes remarkable oxidant stress, does not increase GSH levels in ELF, but several studies have shown that GSH levels in the BAL fluid and ELF of chronic smokers are raised.74–76 On the other hand, GSH is not increased intracellularly—that is, in the BAL fluid cells (mainly alveolar macrophages) of smokers or patients with COPD; thus, extracellular GSH clearly does not reflect intracellular antioxidant enzymes in the lungs of smokers.76 The regulation of ELF GSH levels is still poorly understood; it may be related both to the epithelial injury and also to complex mechanisms of GSH transport and synthesis.
Several studies indicate that GPX 3 is increased during the oxidative stress of human airways and is increased both in the bronchial epithelium and ELF of smokers77 and in asthmatic airways.34 The results with GCL are variable, partly due to the many experimental designs used in animal models and cells.38 Two studies on smokers and/or patients with COPD have shown increased gene expression of GCL in the bronchial epithelium,65,78 while others have reported lowered GCL immunoreactive protein in the bronchial epithelium and alveolar macrophages of subjects with smoker’s lung and COPD.79,76 On the other hand, the extent of GCL immunoreactivity was increased in the metaplastic/dysplastic bronchial lesions of COPD,79,80 as was also the case with some other protective enzymes such as TRX, pointing to the importance of oxidant stress in lung/bronchial epithelial cell carcinogensis.80 The variable results in GCL expression in smoker’s lung and COPD may therefore be related to the cell type present in the epithelium (either normal or metaplastic), and to differences in the smoking history and disease severity or its activity. Induction of antioxidant/detoxification enzymes protects not only normal bronchial epithelium against oxidative insults, but possibly also against cell death/apoptosis, which then might maintain cell proliferation even though the cells contain oxidised DNA alterations.
In addition to the classical antioxidant enzymes, GSTs and heme oxygenase-1 have been investigated in the airways of smokers since it is known that both these enzymes can protect lung cells against oxidants. In one study on a relatively small number of patients, smoking had no effect on GST levels,81 while the transcripts of several GSTs were increased in the bronchial epithelium of smokers without COPD.65 Heme oxygenase-1, an enzyme that is highly inducible by oxidants, is increased in the alveolar macrophages of smokers but decreased in severe COPD.82,83 Moreover, heme oxygenase-1 can be induced, especially in the alveolar macrophages of acute asthmatics.84 There are remarkable differences in the individual variability of the detoxification enzymes (GSTs) and heme oxygenase-1 in human airways, but several studies suggest their induction during the oxidative stress caused by cigarette smoke. Recent results also suggest that, during the ongoing progression of COPD, there is some decline in the efficacy of protective mechanisms in human airways.
Cigarette smoking is also associated with airway/interstitial lung diseases, RB-ILD/DIP as smokers account for 90% of all cases. Several antioxidant enzymes and related mechanisms have been found to be increased in these diseases, probably due to oxidant stress and inflammation. Enzymes that have been found to be increased in these disorders include MnSOD, GCL, TRX, and heme oxygenase-1 (box 1).28,35,47,85
Box 1 Activity and/or expression of antioxidant enzymes, redox modulatory proteins, and detoxification enzymes in smoking related non-malignant lung diseases
Manganese superoxide dismutase (MnSOD)
-
Unchanged gene expression in the airway epithelium of smokers65
-
Increased activity and immunoreactivity in the alveolar epithelium of smokers71
-
Increased immunoreactivity in the inflammatory areas of interstitial lung disorders (DIP)28
CuZn superoxide dismutase (CuZnSOD)
-
Unchanged gene expression, protein and activity in the airway epithelium and lung homogenates of smokers65,71
-
Decreased activity in the alveolar macrophages of elderly smokers70
Extracellular superoxide dismutase (ECSOD)
-
Unchanged gene expression in the airway epithelium of smokers65
-
Unchanged immunoreactivity in the lung homogenates of smokers71
Catalase
-
Unchanged gene expression in the airway epithelium of smokers65
-
Increased catalase activity in the alveolar macrophages of chronic smokers69
Enzymes associated with glutathione metabolism
-
Increased glutamate cysteine ligase (GCL) (gene expression) or decreased (immunoreactive protein) in airway epithelium of smokers65,78,79
-
Increased GCL immunoreactivity in metaplastic and dysplatic epithelium of smokers79,80
-
Increased GCL immunoreactivity in the inflammatory areas of DIP35
-
Increased GPX2, GPX3 and glutathione reductase gene expressions in the bronchial epithelium of smokers65
-
Increased GPX3 gene expression in the bronchial epithelium of smokers113
-
Decreased GPX3 gene expression in severe emphysema66
-
Increased GSTs (gene expressions) in airway epithelium of smokers65
-
Decreased GST and GPX activities in the alveolar macrophages of elderly smokers70
Thioredoxin and thioredoxin peroxidases (peroxiredoxins)
-
Unchanged (thioredoxin gene expression) in the airways of smokers65
-
Increased (thioredoxin reductase gene expression) in the airways of smokers65
-
Increased thioredoxin immunoreactive protein in the inflammatory areas of DIP47
Heme oxygenase-1
-
Increased immunoreactivity in the alveolar walls of smokers83
-
Decreased immunoreactivity in the lung of severe COPD82
-
Increased immunoreactivity in the inflammatory areas of DIP85
Antioxidants and antioxidant enzymes of inflammatory cells and lung inflammation
A typical feature of cigarette smoking related lung diseases such as COPD is the recruitment of macrophages and neutrophils to the lung which increases oxidant burden also in the lung interstitium. Exogenous antioxidants and new synthetic molecules with SOD-like activity decrease lung inflammation in several experimental models of lung injury including cigarette smoke (see next section). However, little is known about the effects of antioxidant enzymes of inflammatory cells themselves on the survival/apoptosis or persistence of inflammation in human lung. Antioxidant enzymes of the inflammatory cells can have a pivotal role in the inflammatory reaction of the lung by offering significant protection against oxidants both intracellularly and extracellularly. They can prolong the survival of inflammatory cells, decrease their apoptosis, but some of them are also released into the interstitium with consequent protection of the extracellular matrix (GSH and extracellular antioxidant enzymes such as ECSOD). Alveolar macrophages have a long life span (several months) and there are suggestions that the survival of macrophages is prolonged in the lungs of smokers and subjects with COPD.86 Macrophages express several antioxidant enzymes and related compounds including at least MnSOD, ECSOD, catalase, enzymes associated with the TRX-PRX system, and especially GRX and HO-1.26,32,47,48,50,87,88 MnSOD and HO-1 appear to be increased in the alveolar macrophages of subjects with smoker’s lung, mild COPD, and acute asthmatic inflammation.83,84 In contrast to alveolar macrophages, the life span of neutrophils is very short. The physiological half life of neutrophils in circulation is only 6 hours, and there is evidence that neutrophil apoptosis is further enhanced by oxidants.89 Very few studies have investigated antioxidant mechanisms of human neutrophils. These studies have shown that typical features of the antioxidant profile of neutrophils are their high catalase content, low levels of GSH-dependent enzymes, and nearly non-detectable levels of TRX and HO-1 by immunohistochemistry.32,87,90 Antioxidant enzymes in the inflammatory cells have been shown to decrease during the progression of several systemic diseases (reviewed by Kinnula90), but whether this also occurs in smoking related human lung diseases or has clinical significance is unclear.
POTENTIAL EXOGENOUS ANTIOXIDANTS AND ANTIOXIDANT MIMETICS IN SMOKING RELATED LUNG DISEASES
Several studies have investigated the systemic oxidant stress which occurs in COPD such as observations of a decline in lung and plasma levels of antioxidants, but variable results have been obtained by administering exogenous antioxidants (mainly vitamins such as vitamin E) to patients with COPD.91 The disappointing results can be explained, at least in part, by the complex cell specific antioxidant systems present in human lung and the fact that vitamins taken orally do not reach the critical areas of the lung where the oxidant stress is highest. Since GSH is considered as one of the most important antioxidants in human lung, numerous studies have tried to improve GSH homeostasis in oxidant mediated lung injury. However, GSH has a poor penetration capability. Neither inhalation nor oral ingestion of GSH have improved the GSH balance in oxidative stress; in fact, it has caused side effects such as bronchoconstriction.74 GSH is readily transported to the extracellular space by several mechanisms (including GSTs and MRPs), but is not directly transported from the extracellular milieu into the cells. N-acetylcysteine (NAC) is a widely used drug that might improve GSH homeostasis. In animal models NAC has attenuated elastase induced emphysema.92 It is relatively safe also in humans and has already been tested in a variety of conditions and treatment schedules, being used orally, by inhalation, and intravenously (reviewed by Rahman74). On the other hand, NAC may also have some pro-oxidant effects at the cellular level. In rats, NAC attenuated pulmonary inflammation caused by various antigens; it also decreased neutrophil recruitment and P-selectin upregulation.93 NAC also decreased H2O2 levels in the exhaled air in COPD and COPD exacerbations.94–97 On the other hand, it had relatively minor effects on lung GSH homeostasis.98–100 Both controlled studies and a systematic review with NAC in COPD have shown a small reduction in exacerbations.101,102 Other compounds with NAC-like activity have been tested in many laboratories including N-acystelyn, GSH esters and GSH precursors, but there are no studies on these compounds in human smokers with COPD. Antioxidants that improve GSH homeostasis have potential possibilities in protecting the lung during oxidative stress, but these need to be investigated in future studies in combination with other new drugs with antioxidant activity.
Another group of antioxidants that have had beneficial results in several animal models are SODs. In the initial experiments, exogenous CuZnSOD or recombinant MnSOD were administered by inhalation or instillation, often encapsulated to liposomes or in combination with other antioxidant enzymes such as catalase (reviewed by Kinnula and Crapo24). However, antioxidant enzymes have several drawbacks such as high immunogenity. In another approach, synthetic SOD mimetics have been developed to avoid the problems associated with endogenous antioxidant enzymes and to improve their penetration capability and efficacy. They have been shown to have at least four properties—namely, scavenging of superoxide, hydrogen peroxide, peroxynitrite, and lipid peroxides. These agents include salen compounds,103 macrocyclics,104 and metalloporphyrins.105 Salen compounds (such as EUKs) can attenuate oxidant injury in numerous organs including lung injury in ARDS.106 The macrocyclic agents (such as M40403) have been reported to decrease lung inflammation and pleurisy by inhibiting ROS/RNS formation, neutrophil infiltration, and release of pro-inflammatory cytokines.104 Metalloporphyrins such as manganese (III) meso-tetrakis (4-benzoic acid) porphyrin (MnTBAP) have been tested in a number of models of lung injury including paraquat and carrageenan-induced pleurisy, exposure to bleomycin, asbestos and radiation, in airway inflammation, and models of bronchopulmonary dysplasia and lung haemorrhage.107–111 In all these models, these synthetic compounds have had very positive results including a significant effect on lung protection. MnTBAP has also been found to reduce smoking related lung injury in rat lungs.112 In that study, one of the compounds, AEOL 10150, significantly decreased the BAL cell number within the first weeks of exposure. Importantly, squamous metaplasia following 8 weeks of tobacco smoke exposure was only 2% in animals treated with the drug compared with 12% in animals exposed to cigarette smoke without AEOL 10150. This study clearly showed that new synthetic SOD mimetics can significantly reduce the adverse effects of exposure to tobacco smoke and suggest that they may even have beneficial effects in preventing malignant conversion of epithelial cells. These new synthetic antioxidants may prove also to have significant importance in human lung inflammation caused by cigarette smoke.
CONCLUSIONS
The lung is exposed to high levels of oxidants, not only in normal conditions but especially in inflammatory states such as cigarette smoking related disorders. The lung has an efficient but complicated antioxidant defence system, where antioxidant enzymes and related detoxification enzymes have a cell specific expression and compartmentalisation and variable induction capacities. Cigarette smoke leads to an induction of some of these enzymes, especially in the bronchial and alveolar epithelium, possibly reflecting high oxidant burden in these locations. Although there is a systemic oxidant stress in COPD, one key determinator in preventing COPD and emphysema progression might be provision of treatment that is specifically directed to the target areas where the oxidant stress is high and the cells are especially vulnerable to injury.
REFERENCES
Footnotes
-
Funded by Helsinki University Hospital, Sigrid Juselius Foundation, Finnish Antituberculosis Association Foundation, and the Cancer Society of Finland.