Article Text

Abstract
Background: Guidelines recommend inhaled corticosteroids (ICS) as maintenance treatment for patients with chronic obstructive pulmonary disease (COPD) with a post-bronchodilator forced expiratory volume in 1 second (FEV1) <50% predicted and frequent exacerbations, although they have only a small preventive effect on the accelerated decline in lung function. Combined treatment with ICS and long acting β2 agonists (LABA) may provide benefit to the stability of COPD, but it is unknown if withdrawal of ICS will result in disease deterioration.
Methods: The effects of 1 year withdrawal of the ICS fluticasone propionate (FP) after a 3 month run-in treatment period with FP combined with the LABA salmeterol (S) (500 μg FP + 50 μg S twice daily; SFC) were investigated in patients with COPD in a randomised, double blind study. 497 patients were enrolled from 39 centres throughout the Netherlands; 373 were randomised and 293 completed the study.
Results: The drop out rate after randomisation was similar in the two groups. Withdrawal of FP resulted in a sustained decrease in FEV1: mean (SE) change from baseline −4.4 (0.9)% (S) v −0.1 (0.9)% (SFC); adjusted difference 4.1 (95% CI 1.6 to 6.6) percentage points (p<0.001). Corresponding figures for the FEV1/FVC ratio were −3.7 (0.8)% (S) v 0.0 (0.8)% (SFC) (p = 0.002). The annual moderate to severe exacerbation rate was 1.6 and 1.3 in the S and SFC groups, respectively (adjusted rate ratio 1.2; 95% CI 0.9 to 1.5; p = 0.15). The mean annual incidence rate of mild exacerbations was 1.3 (S) v 0.6 (SFC), p = 0.020. An immediate and sustained increase in dyspnoea score (scale 0–4; mean difference between groups 0.17 (0.04), p<0.001) and in the percentage of disturbed nights (6 (2) percentage points, p<0.001) occurred after withdrawal of fluticasone.
Conclusions: Withdrawal of FP in COPD patients using SFC resulted in acute and persistent deterioration in lung function and dyspnoea and in an increase in mild exacerbations and percentage of disturbed nights. This study clearly indicates a key role for ICS in the management of COPD as their discontinuation leads to disease deterioration, even under treatment with a LABA.
- COPD, chronic obstructive pulmonary disease
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- LABA, long acting β2 agonists
- MEF50, maximal expiratory flow at 50% of FVC
- PEF, peak expiratory flow
- chronic obstructive pulmonary disease
- exacerbations
- inhaled corticosteroids
- lung function
- symptoms
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- LABA, long acting β2 agonists
- MEF50, maximal expiratory flow at 50% of FVC
- PEF, peak expiratory flow
Chronic obstructive pulmonary disease (COPD) is characterised by progressive airflow limitation that is not fully reversible and that is caused by an abnormal inflammatory response to inhaled noxious particles and gases. COPD has emerged as a worldwide source of morbidity and mortality. In addition to the prevention of disease progression, goals in the management of this disease are to prevent and control symptoms, to reduce the frequency and severity of exacerbations, and to improve health status and exercise tolerance.1 In particular, frequent exacerbations induce a high economic, social, and personal burden.2
Bronchodilator medications are considered central to the symptomatic management of COPD.1 Regular treatment with long acting β2 agonists (LABA) is more effective and convenient than treatment with short acting bronchodilators.3 Inhaled LABA are known to increase the number of symptom-free days in patients with COPD.4,5 Salmeterol in doses of 50 μg twice daily has been shown to improve health status, symptoms, and lung function.3,4,6
Because of the inflammatory nature of COPD, inhaled corticosteroids (ICS) are also used. Their use is recommended by the GOLD and the recent NICE guidelines for patients with a post-bronchodilator forced expiratory volume in 1 second (FEV1) <50% of predicted and frequent exacerbations1,7 since it reduces the number of exacerbations even though it does not clearly modify the long term decline in FEV1 in patients with COPD.1,8–10 A recent meta-analysis, however, suggests a potential role (albeit small) for ICS in modifying the long term natural history of COPD.11 Furthermore, ICS reduce symptoms and the incidence of COPD exacerbations.12,13 Combining ICS and inhaled LABA has provided additive effects over LABA alone on pre-bronchodilator FEV1,14–17 morning peak expiratory flow (PEF),14,18 and symptoms.15
Most available studies on the efficacy of ICS in combination with LABA have been based on step-up study designs,14–18 and little is known about withdrawal of ICS from ICS/LABA treatment on clinical parameters. A study was therefore undertaken to investigate whether the potentially beneficial effects of the ICS fluticasone propionate on FEV1 remain after long term (1 year) withdrawal of ICS following 3 months run-in treatment with the combination of the LABA salmeterol and fluticasone. Secondary end points were lung function, time to first exacerbation, incidence and severity of exacerbations, symptoms, and health status. The run-in period of 3 months was chosen to be on the safe side of the GOLD guidelines which recommend a trial period of 6 weeks to 3 months to get a plateau in the response in FEV1 to ICS.1 This relatively long run-in period probably better mimics daily practice of ICS maintenance therapy.
METHODS
Patients
Patients were recruited from 39 centres in the Netherlands (general and academic hospitals) by their chest physician. Inclusion criteria for entry to the study were: male or female patients aged 40–75 years, an established history of COPD, current or ex-smokers with a smoking history of at least 10 pack years, pre-bronchodilator FEV1 30–70% of predicted, FEV1/FVC ratio <88% for men and <89% for women, and reversibility <10% of predicted normal FEV1 after inhaling 400 μg salbutamol. Furthermore, patients had a history of at least two COPD exacerbations in the last year treated with a course of oral corticosteroids and/or antibiotics.
Patients were excluded if they had respiratory disorders other than COPD, were using regular oxygen therapy, maintenance systemic corticosteroids, or other investigational drugs in the 4 weeks before entry to the run-in period, had serious uncontrolled (psychological) disease, myocardial infarction, acute heart failure, or angina pectoris in the 3 months before entry to the run-in period, were hypersensitive to one of the study drugs, had evidence of alcohol, drug or solvent abuse, or had previously been enrolled to the present study.
Exclusion criteria before entry to the double blind treatment period were use of systemic corticosteroids or antibiotics, hospitalisation for lower respiratory tract infection and/or COPD exacerbation in the run-in period, in which all patients were treated with salmeterol/fluticasone combination treatment. The salmeterol/fluticasone run-in period could be repeated once in case one or more of the above criteria was applicable, starting when the event was resolved.
Approval was obtained from the ethics committees at each participating centre and all patients provided written informed consent.
Study design
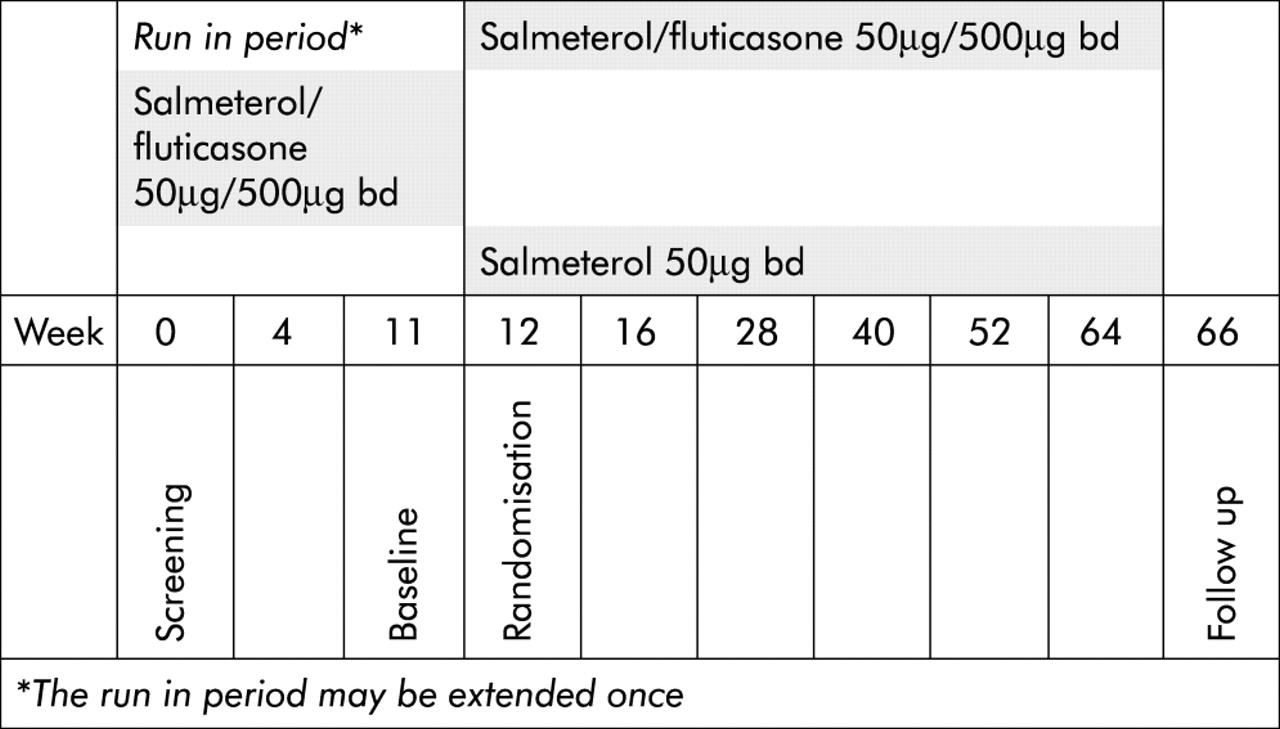
The COSMIC (COPD and Seretide: a Multi-Center Intervention and Characterization) study had a multicentre, randomised, double blind, parallel group design (fig 1). All patients received combined salmeterol 50 μg and fluticasone 500 μg (Seretide 50/500) twice daily (in the morning and evening) via the Diskus inhaler during a 3 month run-in period. Patients were then randomised to 12 months of treatment with either salmeterol/fluticasone or salmeterol alone. A randomisation schedule generated by the patient allocation for clinical trails (PACT) program was used to assign patients to treatment groups. The participating centres received a list of patient (assigned at first visit) and treatment numbers. Patients satisfying the eligibility criteria were assigned the next sequential treatment number from the list. The study medication was packed in identical inhaler devices and labelled to ensure that both the patient and investigator were unaware of the allocated treatment. The centres received hidden entry envelopes with the medication code only to be opened in case of emergencies. Inhaled salbutamol was used as relief medication and anticholinergics and methylxanthines in constant doses were allowed throughout the study. Patients had visits for clinical assessments at weeks 0, 4, 11, 12, 16, 28, 40, 52, 64 and 66 after entering the study.

Study design.
Measurements
Lung function
FEV1, forced vital capacity (FVC), peak expiratory flow (PEF), and maximal expiratory flow at 50% of FVC (MEF50) were calculated from the flow-volume curve using a spirometer (Masterlab, Jaeger, Würzburg, Germany). The Tiffeneau index was calculated as FEV1/FVC. Lung function parameters were expressed as a percentage of reference values.19 After short acting bronchodilating medication was stopped for 6 hours and LABA and study medication for 12 hours, reversibility was assessed by inhaling 400 μg salbutamol using a Volumatic spacer and expressed as a percentage of the predicted FEV1 values. Dynamic lung volumes (FEV1, FVC, PEF, and MEF50) were measured before and 10 minutes after administration of salbutamol.
Exacerbations and rescue medication use
Patients recorded their usage of rescue medication (salbutamol) over the last 24 hours in daily record cards. If a patient on two or more consecutive days used three or more extra inhalations of salbutamol per 24 hours above their reference rescue value (RRV; mean daily salbutamol use in the run-in period), this was counted as one mild exacerbation. A mild exacerbation was considered to have ended if the patient used less than three extra inhalations of salbutamol during at least two consecutive days. The percentage of rescue medication-free days was also used in the analysis. If the patient’s condition worsened and a course of oral corticosteroids was indicated based on a clinician’s judgment (standardised course of prednisolone tablets 30 mg/day for 10 days at the discretion of the physician accompanied by a 10 day course of antibiotics), the exacerbation was defined as moderate. If hospitalisation was required at the discretion of the clinician, the exacerbation was considered severe.
Daily record cards and morning PEF
Every morning patients recorded severity scores of symptoms over the previous 24 hours on dyspnoea, cough, sputum production, and night time sleep disturbance (breathlessness 0 (none) to 4 (breathless at rest); cough and sputum production 0 (none) to 3 (severe); night time sleep disturbance due to respiratory symptoms 0 (none) to 4 (did not sleep at all). A disturbed night was defined as score 1–4. Patients recorded their daily morning PEF throughout the run-in and the treatment periods before inhalation of COPD (study) medication. The highest of three values was used for analysis.
Health status
Disease-specific health status was measured using the St George’s Respiratory Questionnaire (SGRQ).20 A treatment induced change in total score of ⩾4 points is considered clinically significant.21 In addition, the Clinical COPD Questionnaire (CCQ)22 was administered. For both questionnaires a higher score means greater impairment in health status.
Statistical analysis
A power analysis based on data from the TRISTAN study15 showed that 154 evaluable patients per group were required (two sided alpha = 0.05, power 90%) to detect a 100 ml difference in the mean change from baseline in FEV1 after 1 year of treatment. A target enrolment of approximately 515 patients was planned. Considering an estimated dropout rate of 25–30% at the end of the run-in period, this would result in 180 evaluable subjects per treatment group.
The effect on the primary efficacy parameter pre-bronchodilator FEV1 was analysed by repeated measurements analysis of variance (RmANOVA) using SAS PROC MIXED (SAS Institute Inc, Cary, NC, USA) with baseline value at randomisation, age, smoking status, sex, and centre location grouped by geographical area as covariates. The same method was used to compare the secondary end points FVC, PEF, MEF50, and the Tiffeneau index. In these analyses lung function measurements were discarded if the patient had taken short acting bronchodilators <6 hours before the test or LABA or study medication <12 hours before the test.
Exacerbation rates were compared using Poisson regression with adjustment for baseline FEV1 (% predicted), age, sex, smoking status, and centre. Standard errors of estimated effects in this analysis were determined using the bootstrap method. Kaplan-Meier curves and log rank tests were calculated to evaluate the time until the first exacerbation. A Cox’s proportional hazard model, allowing for the same covariates as in the Poisson regression above, was used to evaluate the time until the first severe exacerbation.
From the diary cards the mean daily morning PEF (maximum of three efforts), mean daily recorded number of salbutamol uses in the last 24 hours, and the mean daily scores for dyspnoea, cough, sputum, and night time sleep disturbances were calculated for each inter-visit follow up period. Baseline values for these items were determined from the last 14 days of the run-in period. Only period outcomes which were properly scored by the patient in at least 50% of diary days between visits were considered evaluable and analysed. RmANOVA with the baseline value as covariate was used to evaluate changes in diary card outcomes during treatment. Treatment comparisons for the change from baseline in health status, as measured by the SGRQ and CCQ total scores, were analysed using RmANOVA with baseline value at randomisation, age, sex, centre, and smoking status as covariates. p values of 0.05 (two sided) were considered as the limit of significance in all analyses.
RESULTS
497 patients from 39 centres throughout the Netherlands were enrolled in the 3 month run-in period, of whom 373 were randomised (189 in the salmeterol/fluticasone group and 184 in the salmeterol group), 155 and 138 patients respectively completing the study (fig 2). 49 patients had a repeated run-in because of an exacerbation and 124 were withdrawn, 92 due to an adverse event. Of these 92 withdrawn patients, 78 were excluded because of a moderate to severe COPD exacerbation, 57 of whom were treated with prednisolone. Table 1 shows the demographic data and baseline characteristics of the randomised groups and also some pre-run-in values. There were no relevant differences at baseline between the patients in the salmeterol group and those in the salmeterol/fluticasone group. The drop out rate after randomisation did not differ between the treatment arms but was higher in patients with an FEV1 <50% than in those with an FEV1 ⩾50% (p = 0.006) and in women than in men (p = 0.009).
Demographic data and baseline characteristics of randomised patients
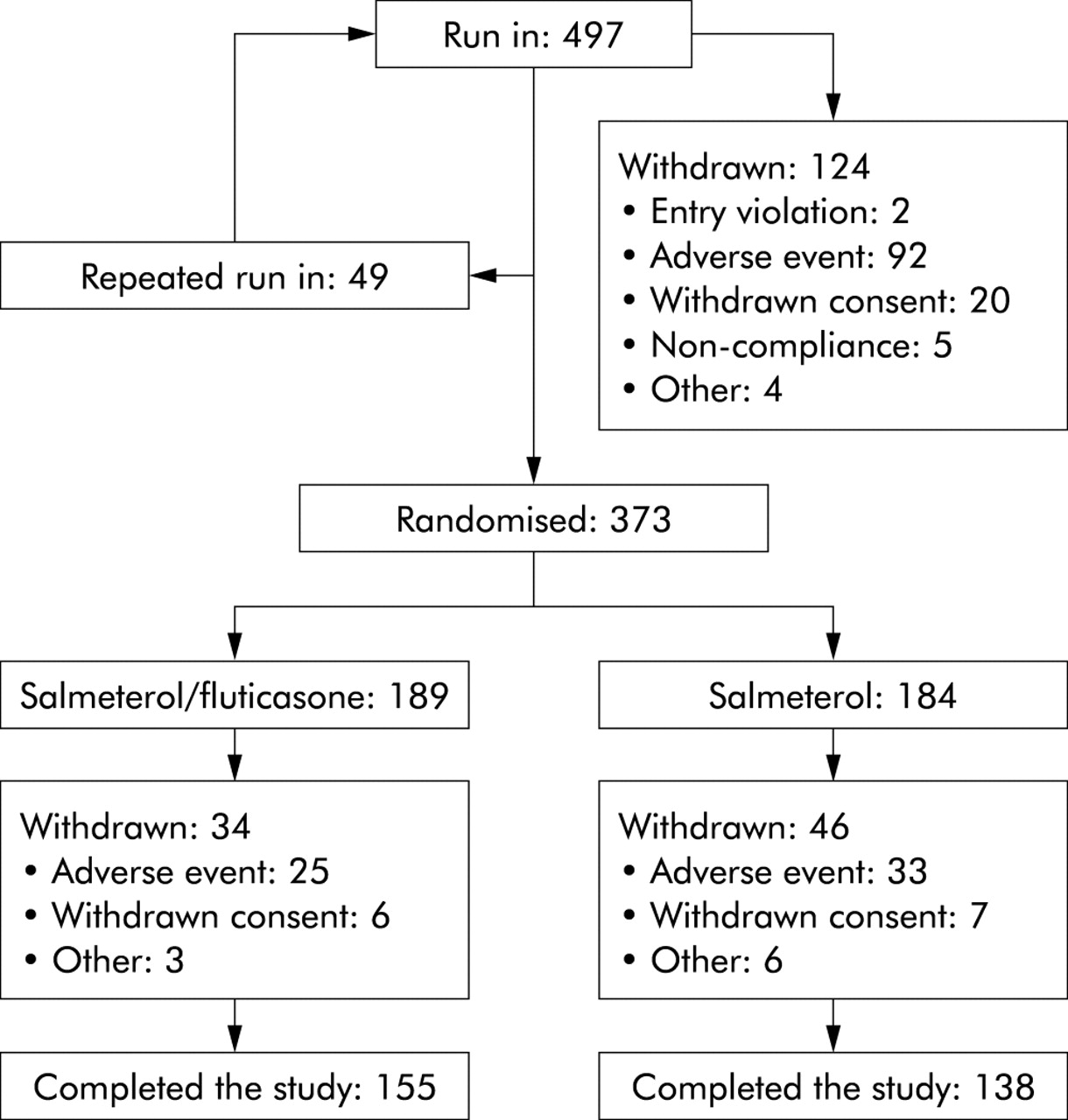
Study profile.
Withdrawal of fluticasone after the run-in period resulted in a prompt and sustained decrease in FEV1 (fig 3A). The mean change at the five visits after randomisation in FEV1, expressed as percentage change from baseline, was −4.4 (0.9)% (salmeterol) v −0.1 (0.9)% (salmeterol/fluticasone). The adjusted difference (salmeterol/fluticasone arm minus salmeterol arm) was 4.1 (95% CI 1.6 to 6.6) percentage points (p<0.001). Within-group comparisons with baseline values at randomisation showed that, in the salmeterol group, the mean FEV1 was significantly reduced at all five visits (all p<0.01). In the salmeterol/fluticasone group only at the 12 month visit was a significant decrease found (p = 0.008). The same applied to the within-group comparisons with the values measured at the start of the run-in period. At the visit 1 year after randomisation the adjusted difference (salmeterol/fluticasone arm minus salmeterol arm) of mean FEV1 was 0.05 l (95% CI 0.01 to 0.10; p = 0.022).

Change from baseline in (A) FEV1 (p<0.001) and (B) Tiffeneau index (FEV1/FVC; p = 0.002). Triangles: salmeterol; squares: salmeterol/fluticasone. Data are presented as mean (SE).
Similar to FEV1, the mean change in the FEV1/FVC ratio after withdrawal of fluticasone was significantly higher than in the salmeterol/fluticasone group (−3.7 (0.8)% (salmeterol) v 0.0 (0.8)% (salmeterol/fluticasone), p = 0.002; fig 3B). The same pattern was observed for PEF (−4.1 (1.0)% (salmeterol) v −0.1 (1.0)% (salmeterol/fluticasone), p = 0.010) and MEF50 (−5.4 (1.8)% (salmeterol) v −0.7 (1.7)% (salmeterol/fluticasone), p = 0.025). The mean change in daily home measured PEF values from the run-in period was significantly greater in the salmeterol/fluticasone group than in the salmeterol group (6.4 (2.8)% v −10.6 (3.0)%, p<0.001).
In 24 patients (12 salmeterol, 12 salmeterol/fluticasone) the non-reversibility requirement (<10%) could not be reliably assessed since at entry into the study they used bronchodilators before lung function testing. However, on retrospective analysis, 15 of these patients had documented non-reversibility in the last year before entry to the study. The remaining nine patients (four salmeterol, five salmeterol/fluticasone) were considered as having a major violation of entry criteria. In addition, three patients (one salmeterol, two salmeterol/fluticasone) were found to have used forbidden medication. Analysing returned study medication, 18 patients (eight salmeterol, 10 salmeterol/fluticasone) had used less than 80% of the prescribed dose and in five patients (three salmeterol, two salmeterol/fluticasone) compliance could not be assessed due to an insufficient number of returned inhalers. These 23 patients were considered to have a severe protocol violation. Altogether, 34 patients (16 salmeterol, 18 salmeterol/fluticasone) were found to have at least one protocol violation. Repeating the analysis for the primary end point with exclusion of these patients for the per-protocol analysis provided similar results as were found in the intention to treat analysis above.
Follow up in the salmeterol and salmeterol/fluticasone group amounted to 161 and 179 person-years, respectively. During this period 109 patients in the salmeterol group experienced a total of 254 exacerbations, of which 19 were severe. In the salmeterol/fluticasone group 115 patients had a total of 238 exacerbations of which 24 were severe. By Poisson regression analysis allowing for treatment, sex, age, baseline FEV1 % predicted and smoking status, the annual moderate to severe exacerbation rate was 1.6 per patient year in the salmeterol group and 1.3 per patient year in the salmeterol/fluticasone group (1.2-fold greater with salmeterol, 95% CI 0.9 to 1.5; p = 0.15). When moderate and severe exacerbations were considered separately, no difference was observed between the treatment arms.
The analysis further showed that women had a 1.4-fold higher exacerbation rate than men (p = 0.03). In addition, the rate of severe exacerbations per year in women was 2.0 times that in men (p = 0.044). Baseline FEV1 was strongly associated with the development of exacerbations; each 10 points lower value of FEV1 (% predicted) led to a 1.2-fold increase in exacerbation rate (p<0.001). When only severe exacerbations were considered, patients with an FEV1 ⩽30% and an FEV1 of 30–49% had an exacerbation rate of 4.9 (p = 0.013) and 3.4 (p = 0.004) times the exacerbation rate in patients with an FEV1 ⩾50%.
The first occurrence of a moderate to severe exacerbation stratified for treatment and baseline FEV1 is shown in fig 4. While fluticasone withdrawal had no effect, patients with FEV1 <50% predicted had a shorter time to first exacerbation than patients with FEV1 ⩾50% predicted.

Time to first moderate or severe exacerbation stratified for FEV1; (A) salmeterol and FEV1 ⩾50% (n = 75); (B) salmeterol/fluticasone and FEV1 ⩾50% (n = 75); (C) salmeterol/fluticasone and FEV1 <50% (n = 114); (D) salmeterol and FEV1 <50% (n = 109). A v B: p = 0.30; C v D: p = 0.57.
When current smokers were analysed separately there was a trend towards more moderate to severe exacerbations after withdrawal of fluticasone (exacerbation rate ratio salmeterol v salmeterol/fluticasone 1.4, p = 0.10). The severe exacerbation rate in current smokers did not differ between treatment groups (p = 0.5). Furthermore, the effect of fluticasone withdrawal did not differ significantly between current and ex-smokers (p = 0.3 and p = 0.5 for moderate to severe and severe exacerbation rates, respectively).
The mean annual incidence rate of mild exacerbations was 1.3 in the salmeterol group and 0.6 for those treated with salmeterol/fluticasone (p = 0.020). The adjusted relative mild exacerbation rate was 2.0 (95% CI 1.1 to 3.5, p = 0.016). In line with this, after withdrawal of fluticasone an immediate and sustained decrease in the percentage of rescue medication-free days was observed (p<0.05; fig 5A). The mean percentage of rescue medication-free days was 47 (2)% in the salmeterol and 53 (2)% in the salmeterol/fluticasone group (p = 0.014).
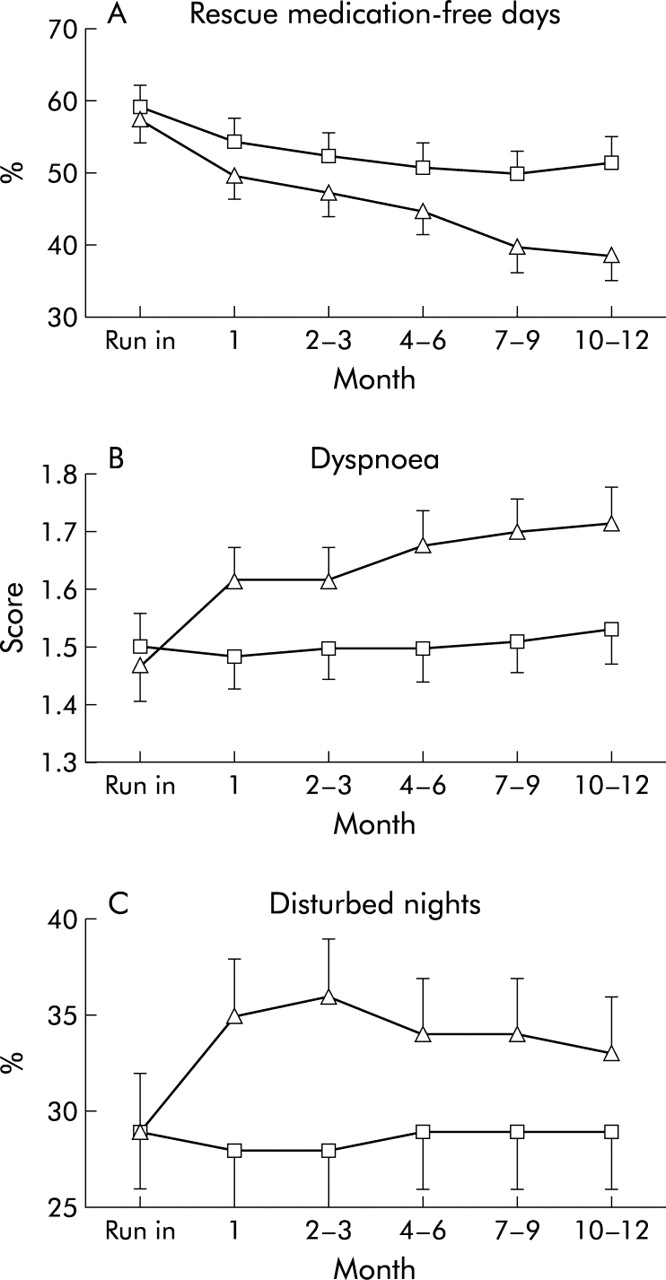
(A) Percentage of rescue medication-free days (p<0.05), (B) dyspnoea score (p<0.001), and (C) percentage of disturbed nights (p<0.001). Triangles: salmeterol; squares: salmeterol/fluticasone. Data are presented as mean (SE).
After withdrawal of fluticasone an increase in the symptom scores for dyspnoea, cough and sputum were observed in the first few days, as well as an increase in the percentage of disturbed nights. The dyspnoea score (mean adjusted difference between the groups 0.17 (0.04), p<0.001; fig 5B) and the percentage of disturbed nights (mean difference between groups 6 (2) percentage points, p<0.001; fig 5C) remained significantly higher throughout the study period. Figure 6A shows that the increase in the cough score was already apparent a few days after withdrawal of fluticasone. However, the difference in cough score and in sputum score between the treatment groups disappeared during follow up with no significant difference over the whole treatment period (fig 6B and C, respectively).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Daily cough score in the first month, (B) cough score (NS) and (C) sputum score (NS). Triangles: salmeterol; squares: salmeterol/fluticasone. Data are presented as mean (SE).
The SGRQ total score increased in both groups, indicating a decrease in health status (salmeterol: from 39.6 (1.1) at baseline to 42.8 (1.3) after 1 year, p<0.001; salmeterol/fluticasone: from 38.6 (1.1) to 41.0 (1.3), p = 0.002). No significant differences were observed between the treatment groups (adjusted difference 0.89 (1.1), p = 0.47). The CCQ total score increased in both groups (salmeterol: from 1.70 (0.06) to 1.89 (0.07), p<0.05; salmeterol/fluticasone: from 1.65 (0.06) to 1.84 (0.07), p<0.05). The CCQ score was significantly higher in the salmeterol group than in the salmeterol/fluticasone groups at months 1 (p = 0.03) and 6 (p = 0.01) but not at 12 months (p = 0.65); the overall adjusted difference between the groups was statistically significant (0.13 (0.06), p = 0.041).
Both treatments were well tolerated; 516 adverse events occurred in the salmeterol treated group and 529 in the salmeterol/fluticasone group (NS). Table 2 summarises the adverse events related to salmeterol or salmeterol/fluticasone treatment as judged by the physicians. There were no differences between the two groups in the number and type of treatment related adverse events.
Treatment related adverse events
DISCUSSION
Guidelines advocate preserving lung function, reducing exacerbations and symptoms, and improving quality of life in the management of COPD with the least possible treatment. This is the first study to investigate the effects of withdrawal of ICS during a 1 year period compared with combined ICS/LABA treatment. This study, with a run-in period in which all patients with moderately severe COPD were treated with a combined dose of salmeterol and fluticasone, shows that withdrawal of fluticasone results in persistent deterioration in lung function, dyspnoea, and percentage of disturbed nights, an increase in mild exacerbations, and a decrease in the percentage of rescue medication-free days despite the continued use of salmeterol.
Discontinuation of fluticasone after the 3 month run-in period with the combination product salmeterol/fluticasone resulted in a prompt and sustained deterioration in FEV1, FEV1/FVC ratio, (daily) PEF, and MEF50. In the combination group lung function parameters remained stable over the 1 year treatment period. Our results are complementary to the previously observed sustained increase in FEV1 in COPD for 1 year15 and for 24 weeks16 after institution of salmeterol/fluticasone treatment (50 μg/500 μg) compared with salmeterol. Hanania et al14 showed that 24 weeks of treatment with salmeterol/fluticasone in a lower dose (50 μg/250 μg) also increased pre-bronchodilator FEV1 and daily PEF compared with salmeterol alone. Szafranski et al18, however, reported no additional effect on FEV1 of 12 months treatment with budesonide/formoterol compared with formoterol alone. In contrast, after a 2 week run-in period with formoterol and 30 mg oral prednisolone, the combination of budesonide/formoterol 320/9 μg resulted in a small but significantly higher improvement in FEV1 (5%) than formoterol alone.17 Differences between the studies can be explained either by the treatment doses used, patients under study, study design (run-in period), or differences in effect between the two drug regimens applied. However, overall, the available data suggest that a combination of ICS and LABA in patients with COPD results in improved lung function and withdrawal of ICS in those using ICS/LABA leads to a deterioration in lung function.
Withdrawal of fluticasone was also clearly associated with an increased number of mild exacerbations and a lower number of rescue medication-free days. Reports in the literature on combined ICS and LABA are variable, with some studies showing a reduction in exacerbations compared with placebo or LABA alone,14,15,18 and others not reporting this finding.16,17 Our study showed that the overall rate and time to the first moderate or severe exacerbation were not significantly different between the combined ICS and LABA group and the LABA group, a finding compatible with the results in the TRISTAN study.15 This does not, however, imply that these drugs have no effect on the rate of exacerbations per se. Salmeterol alone and combined salmeterol and fluticasone have clearly positive effects when compared with placebo,15 but our study design did not include a placebo arm. Furthermore, the present study was powered to detect an effect on FEV1 and might have been underpowered to distinguish differences in exacerbation rate. In addition, our definition of exacerbations was based on medication use and not on diary card records which may result in a higher exacerbation frequency as, for instance, in the East London study.23
Interestingly, the time to the first moderate or severe exacerbation was significantly shorter in patients with a worse FEV1. This was in line with a sub-analysis of the ISOLDE study in which fluticasone was predominantly effective in decreasing exacerbations in patients with an FEV1 <50%.24 The higher incidence of severe exacerbations found by post hoc analysis in women deserves further research but is compatible with the study by Prescott et al25 who reported more hospital admissions in women with COPD independently of FEV1. Although it is suggested that smoking can attenuate the effects of ICS, we could only identify a trend towards more moderate to severe exacerbations after withdrawal of fluticasone in the subgroup of current smokers.
All assessed symptoms—cough, sputum production, dyspnoea, and the percentage of disturbed nights—increased during the first few days after withdrawal of fluticasone. This immediate effect on symptoms has also been reported when initiation of inhaled treatment with budesonide/formoterol and formoterol were compared—in this case with a better improvement with budesonide/formoterol.18 Thus, patients immediately perceive the institution and withdrawal of inhaled steroids, possibly due to their vasoconstrictor effect resulting in less and more airway wall oedema, respectively.26 However, not all symptoms changed persistently. Worsening of dyspnoea and—possibly as a consequence—an increasing percentage of disturbed nights were sustained after discontinuation of fluticasone for the whole 1 year treatment period. In contrast, the differences between treatments which occurred immediately with regard to sputum and cough disappeared over time, as has been reported in other studies,15,18 possibly indicating adaptation of these symptoms to ICS with time. This may be explained by the different pathology underlying cough and sputum versus dyspnoea. Cough and sputum are airway based and not only inflammation based symptoms that perhaps should be treated with medication in addition to anti-inflammatory treatment to produce a longer term effect than with LABA alone. We realise that the present analyses on symptom scores must be considered as a post hoc descriptive report since the underlying mechanisms of the symptomatology in COPD have not yet been elucidated and clearly require further studies.
Both treatment groups showed a significant deterioration in health status after 1 year, which is similar to the findings in the ISOLDE study.10 The decline in health status measured by the SGRQ did not differ between the groups, however, despite the better FEV1, lower incidence of respiratory symptoms, and higher percentage of rescue medication-free days with the combination treatment. Although at first this appears to be incompatible with the findings of previous studies, this is comparable with studies which did not show a difference between ICS and LABA on SGRQ.15,18 Although SGRQ and CCQ are highly correlated,22 there was an overall positive effect of salmeterol/fluticasone relative to salmeterol alone on clinical control as assessed by the CCQ, a simple tool to assess activity limitation and emotional dysfunction of the patient.22
To date, only three studies have reported results on discontinuation of ICS in COPD. In an observational substudy of the ISOLDE trial, 38% of patients had an exacerbation after withdrawal of ICS treatment compared with 6% of patients not previously treated with ICS.27 A crossover study with a short follow up period of 6 weeks showed that withdrawal of beclomethasone led to deterioration in lung function and exercise induced dyspnoea.28 However, the sample size was small (n = 24) with a wide range of FEV1 values, so the results should be interpreted with caution. Finally, the COPE study showed that withdrawal of fluticasone was associated with a more rapid onset and a higher risk of recurrence of exacerbations and a significant deterioration in health status in 244 patients with COPD.29 The follow up period of this study was relatively short (26 weeks), there was no uniform run-in period with regard to LABA, no controlled additional LABA was given, and no medication based criteria for exacerbations were defined.29 This indicates the unique design and strength of the present study in which we have assessed the effects of fluticasone withdrawal during 1 year in a large group of patients with COPD after a run-in period of 3 months with combined salmeterol and fluticasone treatment.
The drop out rate in our study did not differ between the treatment arms owing to the similar (mainly respiratory) adverse event rate, in line with the studies by Hanania et al14 and Szafranski et al.18 In the TRISTAN study, however, the drop out rate was higher after salmeterol alone than with salmeterol/fluticasone despite similar exacerbation rates in the two groups.15 We found a higher drop out rate in patients with an FEV1 of <50% than in those with an FEV1 of ⩾50% and in women than in men, indicating that women and patients with more pronounced airflow limitation were more prone to (respiratory) adverse events.
Only patients with at least two COPD exacerbations in the past year were included, in theory reducing the generalisability of the study. However, a comparable exacerbation rate was reported earlier in patients with a similar FEV1 (% predicted).30 Furthermore, our study supports the recent NICE/BTS COPD guidelines which recommend that patients with a history of previous exacerbations should be treated with ICS.
Both treatments were well tolerated and there were no differences in adverse events between the groups. Also no differences were observed in adverse events that were judged by the physicians to be related to salmeterol or salmeterol/fluticasone treatment, indicating the safety of the combination therapy.
In conclusion, withdrawal of fluticasone in patients with COPD results in persistent deterioration in lung function, an increase in mild exacerbations, dyspnoea and percentage of disturbed nights, and in a decrease in the percentage of rescue medication-free days and worsening of the CCQ despite the continued use of salmeterol. This study clearly demonstrates a key role for ICS in the management of COPD, as their discontinuation leads to deterioration even under an umbrella of treatment with a LABA.
REFERENCES
Footnotes
-
↵† Deceased.
-
COSMIC investigators: Dr R Aalbers, Dr F Beaumont, Dr W Boersma, Prof Dr J Bogaard, M Bunnik, J Creemers, W Dalinghaus, C de Graaff, Dr J de Jong, Dr P de Jong, D de Munck, D de Vries, I de Vries, W den Hertog, H Dik, E Dubois, M Eland, W Evers, S Gans, W Geraedts, Dr H Heijerman, A Hendriks, Dr Ho, Dr B Hol, J Kersbergen, H Los, P Luursema, B Pannekoek, Dr W Pieters, R Quanjel, E Quanjel-Wisselo, R Rammeloo, J Retera, Dr A Roldaan, Dr A Rudolphus, L Sala, N Schlösser, Dr A Schols, Dr A Schreurs, Dr J Simons, A Sips, Dr F Smeenk, W Strankinga, I Utama, F van Beek, H van de Woude, P van den Berg, Dr J van den Bosch, J van den Bosch, W van der Brink, Dr F van den Elshout, Dr B van der Bruggen- Bogaarts, J van der Zeijden, A van Harreveld, Dr A van Keimpema, Dr J van Noord, H van Pagée, R van Snippenburg, P van Spiegel, Dr J Verbraecken, J Westbroek.E F M Wouters and D S Postma designed the study, analysed the data, reviewed and interpreted the results and wrote the manuscript. B Fokkens† designed the study. W C J Hop performed the statistical analysis of the data and wrote the manuscript. J Prins, A F Kuipers, H R Pasma and C A J Hensing reviewed and interpreted the results. E C Creutzberg analysed the data, reviewed and interpreted the results and wrote the manuscript. All authors have access to all data in the study and held final responsibility for the decision to submit for publication.
-
GlaxoSmithKline provided funding for the study (protocol number SER9602).
-
The study sponsor, GlaxoSmithKline, was involved in the study design, together with the principal investigators, in the collection and analysis of data, which were made freely available to the investigators, and in the decision to submit the paper for publication.
Conflict of interest: E F M Wouters, D S Postma and E C Creutzberg have been consultants for and received research grants from GlaxoSmithKline. B Fokkens† was an employee of GlaxoSmithKline. W C J Hop is a statistical consultant for GlaxoSmithKline. J Prins, A F Kuipers, H R Pasma and C A J Hensing received research grants from GlaxoSmithKline.
Linked Articles
- Airwaves