Article Text

Abstract
Background: Ongoing inflammatory processes resulting in airway and vascular remodelling characterise chronic obstructive pulmonary disease (COPD). Vascular endothelial growth factor (VEGF) and its receptors VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) could play a role in tissue remodelling and angiogenesis in COPD.
Methods: The cellular expression pattern of VEGF, Flt-1, and KDR/Flk-1 was examined by immunohistochemistry in central and peripheral lung tissues obtained from ex-smokers with COPD (forced expiratory volume in 1 second (FEV1) <75% predicted; n = 14) or without COPD (FEV1 >85% predicted; n = 14). The immunohistochemical staining of each molecule was quantified using a visual scoring method with grades ranging from 0 (no) to 3 (intense).
Results: VEGF, Flt-1, and KDR/Flk-1 immunostaining was localised in vascular and airway smooth muscle (VSM and ASM) cells, bronchial, bronchiolar and alveolar epithelium, and macrophages. Pulmonary endothelial cells expressed Flt-1 and KDR/Flk-1 abundantly but not VEGF. Bronchial VEGF expression was higher in microvascular VSM cells and ASM cells of patients with COPD than in patients without COPD (1.7 and 1.6-fold, p<0.01, respectively). VEGF expression in intimal and medial VSM (1.7 and 1.3-fold, p<0.05) of peripheral pulmonary arteries associated with the bronchiolar airways was more intense in COPD, as was VEGF expression in the small pulmonary vessels in the alveolar region (1.5 and 1.7-fold, p<0.02). In patients with COPD, KDR/Flk-1 expression was enhanced in endothelial cells and in intimal and medial VSM (1.3, 1.9 and 1.5-fold, p<0.02) while endothelial Flt-1 expression was 1.7 times higher (p<0.03). VEGF expression was significantly increased in bronchiolar and alveolar epithelium as well as in bronchiolar macrophages (1.5-fold, p<0.001). The expression of VEGF in bronchial VSM and mucosal microvessels as well as bronchiolar epithelium was inversely correlated with FEV1 (r<−0.45; p<0.01).
Conclusions: VEGF and its receptors Flt-1 and KDR/Flk-1 may be involved in peripheral vascular and airway remodelling processes in an autocrine and/or paracrine manner. This system may also be associated with epithelial cell viability during airway wall remodelling in COPD.
- ASM, airway smooth muscle
- COPD, chronic obstructive pulmonary disease
- FEV1, forced expiratory volume in 1 second
- TGF-β1, transforming growth factor β1
- VEGF, vascular endothelial growth factor
- VSM, vascular smooth muscle
- chronic obstructive pulmonary disease
- vascular endothelial growth factor
- angiogenesis
Statistics from Altmetric.com
- ASM, airway smooth muscle
- COPD, chronic obstructive pulmonary disease
- FEV1, forced expiratory volume in 1 second
- TGF-β1, transforming growth factor β1
- VEGF, vascular endothelial growth factor
- VSM, vascular smooth muscle
Chronic obstructive pulmonary disease (COPD) is a disease state characterised by airflow limitation that is not fully reversible, usually progressive, and associated with an abnormal inflammatory response of the lungs in response to noxious particles and gases.1 COPD is a major health problem with cigarette smoking as its main cause. One important pathological feature of COPD is chronic airway inflammation characterised by an influx of inflammatory cells—predominantly neutrophils, macrophages and CD8+ T lymphocytes—in the lumen and wall of the bronchial and bronchiolar airways and parenchyma.2–4 Furthermore, several studies have reported a thickened bronchiolar wall and airway remodelling with peribronchiolar fibrosis, an increase in airway smooth muscle (ASM) mass, and emphysema.3,5,6
Vascular abnormalities have been associated with the development of COPD.7,8 Wright et al found an increase in wall area of small (<500 μm) pulmonary vessels by intimal thickening in patients with mild to moderate COPD and medial thickening in severe cases as well, which was correlated with a decline in forced expiratory volume in 1 second (FEV1).7,9 Furthermore, recent observations have indicated that muscular pulmonary and bronchiolar arteries have increased adventitial infiltration of CD8+ T lymphocytes and have intimal thickening that is correlated with the amount of total collagen deposition.8,10 In addition, emphysema may lead to loss of the pulmonary vascular bed and induce angiogenesis.11 Little is known, however, about the molecular mechanisms underlying these processes in the context of COPD.
One of the potent proteins involved in vascular remodelling is vascular endothelial growth factor (VEGF). The VEGF family currently comprises six members (VEGF-A to F), of which the originally identified VEGF-A165 variant is the predominant form of five additional spliced variants.12 VEGFs are heparin binding proteins and act via their high affinity transmembrane receptors VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1). The receptors belong to the family of tyrosine kinases and are predominantly expressed by endothelial and epithelial cells.12 VEGF promotes an array of responses in the endothelium including hyperpermeability, endothelial cell proliferation, and angiogenesis with new vessel tube formation in vivo.12,13 VEGF expression can be induced under a number of pathophysiological conditions including pulmonary hypoxia and pulmonary hypertension with increased sheer stress.13,14 Both hypoxia and pulmonary hypertension are pathological features often seen in patients with advanced COPD.2 We hypothesise that increased VEGF expression, perhaps under the influence of hypoxia inducible transcription factors, may contribute to increased and abnormal proliferation of endothelial and vascular smooth muscle (VSM) cells in pulmonary vessels leading to vascular remodelling.
Although the role of VEGF in the vascular biology has been studied thoroughly, it has become clear that VEGF and its receptor system are also involved in various other cellular events including epithelial proliferation and survival and the recruitment of mast cells, neutrophils, and macrophages to sites of fibrosis.13,15,16 Recent studies indicate that VEGF is expressed in the lung by bronchiolar, submucosal, glandular and alveolar type I and II epithelial cells, alveolar macrophages, ASM and VSM cells, as well as myofibroblasts in fibrotic lung lesions.14,17,18
In order to assess the role of VEGF and its receptors VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) in the pathophysiology of COPD, we first examined the expression of VEGF-A, Flt-1, and KDR/Flk-1 in central and peripheral lung tissue from ex-smokers with or without COPD. We also investigated the relationship of lung function with the expression of VEGF and its receptors.
METHODS
Selection of patients
Central and peripheral lung tissues were obtained from current or ex-smokers undergoing lobectomy or pneumonectomy for lung cancer. Fourteen subjects with COPD (FEV1 <75% predicted) and 14 subjects without COPD (FEV1 >84% predicted) were included as previously described.19–21,23 Total lung capacities (TLC) were not below normal levels (TLC >80% predicted). None of the patients had an upper respiratory tract infection and they did not receive antibiotics perioperatively. No patients had received glucocorticosteroids during the 3 month period before resection; four patients received glucocorticosteroids perioperatively. Based on all these criteria, subjects with COPD could not be subdivided into patients with either chronic bronchitis or emphysema alone. Clinical data are given in table 1. Subjects were excluded if the obstruction of the central bronchi was due to the tumour, if diffuse pulmonary inflammation or fibrosis was present, or if no tissue free from tumour could be obtained. The patients in the two groups had participated in a larger research project, part of which has been published previously.19–21 The medical ethics committee of LUMC approved the study.
Clinical characteristics of study subjects
The lung tissue specimens used in this study were obtained from the archival collection at the Department of Pathology (LUMC, Leiden, NL). The specimens were routinely fixed in 10% neutral buffered formalin by inflation-immersion fixation and embedded in paraffin for histopathological examination and immunohistochemistry.
Immunohistochemistry
Paraffin sections (4 μm thick) of the lung tissues were cut and mounted on silane coated glass slides. Immunohistochemistry was performed using a method described elsewhere.20,22,23 Briefly, after deparaffinisation in xylene and rehydration through graded alcohol, the slides were rinsed with phosphate buffered saline (PBS). Endogenous peroxidase was blocked with 0.3% hydrogen peroxidase. For VEGF, VEGFR-1, VEGFR-2 and Ki-67 staining, slides were pretreated by boiling in citrate buffer (10 mM, pH = 6.0) for 10 minutes in a microwave oven. The sections were preincubated with 10% normal goat serum diluted in 5% bovine serum albumin (BSA) in PBS (pH = 7.4) and then incubated for 30 minutes at room temperature with affinity purified rabbit polyclonal VEGF antibody in a dilution of 1:200 v/v. The VEGF antibody used was raised against a 20 amino acid synthetic peptide corresponding to residues 1–20 of the amino terminus of human VEGF (Santa Cruz Biotechnology Inc, Santa Cruz, CA, USA). A different series of slides was incubated with a rabbit polyclonal antibody against a synthetic peptide corresponding to aa 1312–1328 of human Flt-1 (Neomarkers RB-1526, Fremont, CA, USA) in a dilution of 1:100 v/v. For VEGFR-2, a rabbit polyclonal antibody against amino acids 1326–1345 of mouse KDR/Flk-1 (Neomarkers RB-1527) in a dilution of 1:200 v/v was used. To examine proliferation of cells in the airways an antibody against Ki-67 (Dako Corporation, Glostrup, Denmark) of 1:400 v/v at 4oC overnight was used as a marker. Consecutive tissue sections were also stained with a monoclonal mouse anti-human alpha-smooth muscle actin (α-SMA) antibody (clone 1A4; Biogenex, San Ramon, USA) in a dilution of 1:1000 v/v. The optimal dilution of the first antibody was identified by examining the intensity of staining obtained with a series of dilutions of the antibody from 1:50 to 1:1000. Negative controls were prepared by omission of the primary antibody.
After washing with Tris-base buffered saline (TBS, pH = 7.4) the test and control slides were incubated for 15 minutes with Powervision post-antibody blocking solution (Immunovision Technologies, Daly City, CA, USA). The slides were then washed and incubated with Powervision polymerised horseradish peroxidase conjugates (Immunovision Technologies) and the sections were stained with 3,3′-diaminobenzidine tetrahydrochloride (Sigma, Zwijndrecht, NL) as chromogen, counterstained with Mayer’s haematoxylin, and visualised with light microscopy.
Quantitative scoring analysis of immunohistochemistry
Before screening, sections were coded so that the observers were unaware of the clinical details of the case under study. Expression of VEGF, Flt-1, and KDR/Flk-1 was analysed semiquantitatively using a visual scoring method with grades ranging from 0 to 3 (0 = no staining; 1 = moderate staining; 2 = intense staining; 3 = very intense staining) as previously described.8,19,20,23 The entire section of a tissue block was investigated and scored at the same magnification. The staining intensity of VEGF, Flt-1, and KDR/Flk-1 in the bronchial and bronchiolar airways as well as the alveolar parenchyma was scored blindly by two independent observers who were unaware of the clinical data of the case under study. Errors within and between observers were examined by correlating the expression scores using Pearson’s analysis and a very high correlation ranging from 0.8 to 0.9 was obtained.
In the bronchial airways staining was assessed in the bronchial epithelium, mucosal microvasculature, submucosal bronchial wall vessels, ASM cells, and macrophages in the bronchial airway wall. In peripheral lung tissues the staining of VEGF and receptors was analysed in the bronchiolar and alveolar epithelium, bronchiolar ASM cells, and bronchiolar and alveolar macrophages. The vasculature in the peripheral lung was further subdivided into the larger pulmonary vessels associated with the bronchiolar airways and smaller vessels situated within the alveolar parenchyma. In each the VEGF and receptor staining of endothelial, intimal, and medial VSM cells was assessed. Since transforming growth factor-β1 (TGF-β1) may also induce VEGF expression in epithelial cells,25,26 we examined the correlation between the epithelial VEGF expression in this study and the epithelial TGF-β1 expression from one of our previous studies.20 In both studies the same patient groups were used and the staining was performed on adjacent or near sections.
Statistical analysis
Data were analysed for statistical significance using the unpaired two-tailed Student’s t test and the non-parametric Mann-Whitney test where appropriate. Data on the expression of VEGF and its receptors were expressed as mean (SE). The staining of VEGF and its receptors for different compartments was correlated with FEV1 using Pearson’s correlation analysis. Differences with p values of ⩽0.05 were considered statistically significant.
RESULTS
Clinical characteristics
The clinical and lung function characteristics of all subjects included in the study are shown in table 1. As defined, the COPD group had decreased FEV1 and FEV1/FVC values (p<0.001), as has been described previously.19–23 The subjects in the two groups did not differ significantly in age and smoking status (pack years) or steroid use.
Immunolocalisation of VEGF, Flt-1, and KDR/Flk-1
Bronchial airways
Examples of VEGF expression in the central airways of non-COPD and COPD subjects are shown in fig 1A and B, while fig 1C and D (both taken from COPD subjects) show the VEGF receptors and KDR/Flk-1, Flt-1 respectively. Within the airways, VEGF, KDR/Flk-1 and Flt-1 were localised in the bronchial epithelium and ASM cells, bronchial microvasculature of the mucosa and submucosa, and on inflammatory cells (predominantly macrophages) in all subjects (fig 1A–D). In the vessel wall, VSM cells were positive for VEGF, Flt-1, and KDR/Flk-1 while endothelial cells did not stain for VEGF protein but were positive for Flt-1 and KDR/Flk-1 (fig 1).

Immunohistochemical localisation of (A, B) VEGF, (C) KDR/Flk-1, and (D) Flt-1 in bronchial tissues from non-COPD (ex-) smoking subjects (A) and patients with COPD (B, C, D). Immunoreactive VEGF, KDR/Flk-1, and Flt-1 were localised in bronchial epithelial cells, airway smooth muscle (ASM) cells, and in macrophages, endothelial and vascular smooth muscle (VSM) cells. Colour was developed with 3,3-diaminobenzidine tetrahydrochloride (DAB) as chromogen (brown colour) and counterstained with Mayer’s haematoxylin. Arrows indicate sites of positivity for VEGF, Flt-1 or KDR/Flk-1. Original magnification ×100; scale bar = 50 μm.
To assess the intensities of VEGF, Flt-1, and KDR/Flk-1 expression in various bronchial airway compartments, a visual scoring method was used as previously described.8,19,20,23 VEGF expression was higher in the bronchial ASM cells of COPD patients than in those of non-COPD subjects (1.6-fold, p<0.01) but not in bronchial epithelial cells and macrophages (fig 2A). In the central airways of patients with COPD, VEGF staining was more intense in the VSM microvasculature of the bronchial mucosal (lamina propria) (1.7-fold, p<0.001) and bronchial VSM in the submucosa (1.4-fold, p<0.01, fig 2A) than in non-COPD subjects. No significant differences were observed in the levels of expression of KDR/Flk-1 and Flt-1 between COPD and non-COPD subjects (fig 2B and C, respectively). In all subjects VEGFR-2 (KDR/Flk-1) expression was more intense than VEGFR-1 (Flt-1) expression, except in the endothelial cells of bronchial microvessels and on bronchial macrophages which were comparable (fig 2B and C).

Expression of (A) VEGF, (B) KDR/Flk-1 and (C) Flt-1 protein in different cell types in bronchial airways using visual scoring. The immunostaining score ranges from 0 (no staining) to 3 (very intense staining). Open and closed bars represent mean data from subjects without and with COPD, respectively. Data are presented as mean (SE). Asterisk indicates a significant difference (p<0.05, Student’s t test) compared with non-COPD subjects. Epi, bronchial epithelium; MV, bronchial microvessels in the mucosa; VSM bron, bronchial vascular smooth muscle cells in the submucosa; ASM, airway smooth muscle; Mφ, macrophages.
Bronchiolar airways
Figure 3 shows peripheral lung tissue from non-COPD and COPD subjects stained for VEGF (fig 3A and B), KDR/Flk-1 (fig 3C and D), and Flt-1 (fig 3E and F). In bronchiolar epithelial cells VEGF (1.5-fold, p<0.001, fig 4A) and Flt-1 expression (1.4-fold, p<0.04, fig 4C) were increased in COPD patients compared with non-COPD subjects whereas the staining for KDR/Flk-1 was unchanged in both patient groups (fig 4B). ASM cells showed slightly increased VEGF expression in the bronchiolar region (1.3-fold, p<0.05), whereas the expression of both the receptors remained unchanged in the two patient groups. However, the expression of KDR/Flk-1 was more intense than Flt-1 in all patients (fig 4B and C). The increase in the expression of VEGF in the larger pulmonary arteries associated with the bronchiolar airways in the intimal and medial VSM cells was 1.7-fold and 1.3-fold, respectively (p<0.05, fig 4A) for COPD and control subjects, while endothelial cells did not express VEGF. KDR/Flk-1 expression was enhanced in endothelial cells and intimal and medial VSM cells (1.3, 1.9 and 1.5-fold, respectively, p<0.02, fig 4B); the corresponding value in endothelial cells for Flt-1 expression was 1.7-fold (p<0.03, fig 4C). In both patient groups the intimal VSM cells stained 2–3 times less intensely than medial VSM cells for VEGF, Flt-1, and KDR/Flk-1. Moreover, vascular Flt-1 expression was lower than KDR/Flk-1 and VEGF expression in each of the investigated vessel wall areas (p<0.002, fig 4). VEGF staining in bronchiolar macrophages (1.5-fold, p<0.001, fig 4A) was higher in COPD subjects than in non-COPD subjects, whereas the staining on macrophages of Flt-1 or KDR/Flk-1 expression in bronchiolar airways as well as VEGF, Flt-1, or KDR/Flk-1 in the alveolar region remained unchanged (fig 5).

Immunohistochemical localisation of (A, B) VEGF, (C, D) KDR/Flk-1, and (E, F) Flt-1 in peripheral tissues from non-COPD (ex-) smoking subjects (A, C, E) and patients with COPD (B, D, F). Immunoreactive VEGF, Flt-1, and KDR/Flk-1 were localised in bronchiolar and alveolar epithelial cells, airway smooth muscle (ASM) cells, macrophages, and in endothelial and intimal/medial vascular smooth muscle (VSM) cells. Colour was developed with 3,3-diaminobenzidine tetrahydrochloride (DAB) as chromogen (brown colour) and counterstained with Mayer’s haematoxylin. Arrows indicate sites of positivity for VEGF, Flt-1, or KDR/Flk-1. Original magnification ×100; scale bar = 50 μm.

Expression of (A) VEGF, (B) KDR/Flk-1, and (C) Flt-1 protein in different cell types in bronchiolar airways and associated pulmonary arteries using visual scoring. Open and closed bars represent mean data from subjects without and with COPD, respectively. Data are presented as mean (SE). Asterisk indicates a significant difference (p<0.05, Student’s t test) compared with non-COPD subjects. Epi, bronchiolar epithelium; EC, endothelial cells; VSM int, VSM med, intimal and medial vascular smooth muscle cells; ASM, airway smooth muscle cells; Mφ, bronchiolar macrophages.
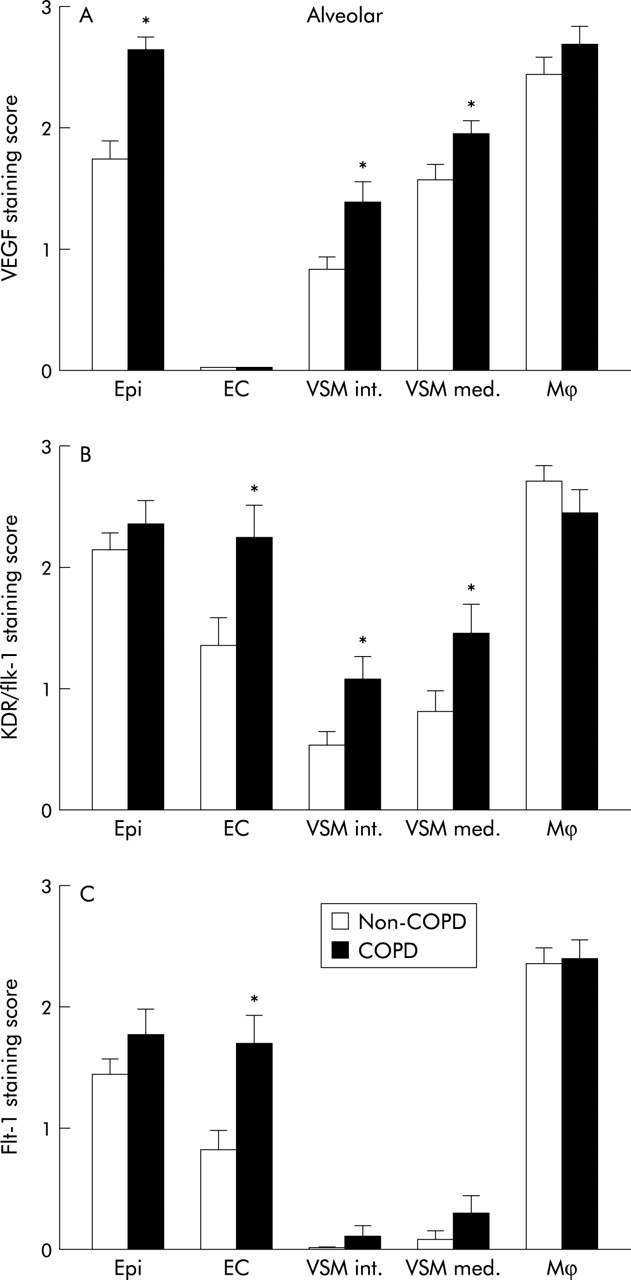
Expression of (A) VEGF, (B) KDR/Flk-1, and (C) Flt-1 protein in different cell types in alveolar parenchyma and pulmonary vasculature using visual scoring. Open and closed bars represent mean data from subjects without and with COPD, respectively. Data are presented as mean (SE). Asterisk indicates a significant difference (p<0.05, Student’s t test) compared with non-COPD subjects. Epi, bronchiolar epithelium; EC, endothelial cells; VSM int, VSM med, intimal and medial vascular smooth muscle cells; Mφ, bronchiolar macrophages.
Alveolar parenchyma
Staining of alveolar epithelial cells (type I and II) for COPD was more intense than for non-COPD controls (1.5-fold, p<0.0001, fig 5A). KDR/Flk-1 and Flt-1 expression were not changed in alveolar epithelial cells (fig 5B and C). VEGF expression was increased in intimal and medial VSM cells (1.5 and 1.7-fold, p<0.01, fig 5A) of small pulmonary vessels in the alveolar region; the corresponding values for KDR/Flk-1 were 2.0 and 1.8-fold, respectively (p<0.02, fig 5B). The expression of both KDR/Flk-1 and Flt-1 was increased in endothelial cells of small pulmonary vessels in the lung parenchyma (1.7 and 2.1-fold, respectively, p<0.001, fig 5B and C).
Correlation between staining and clinical data
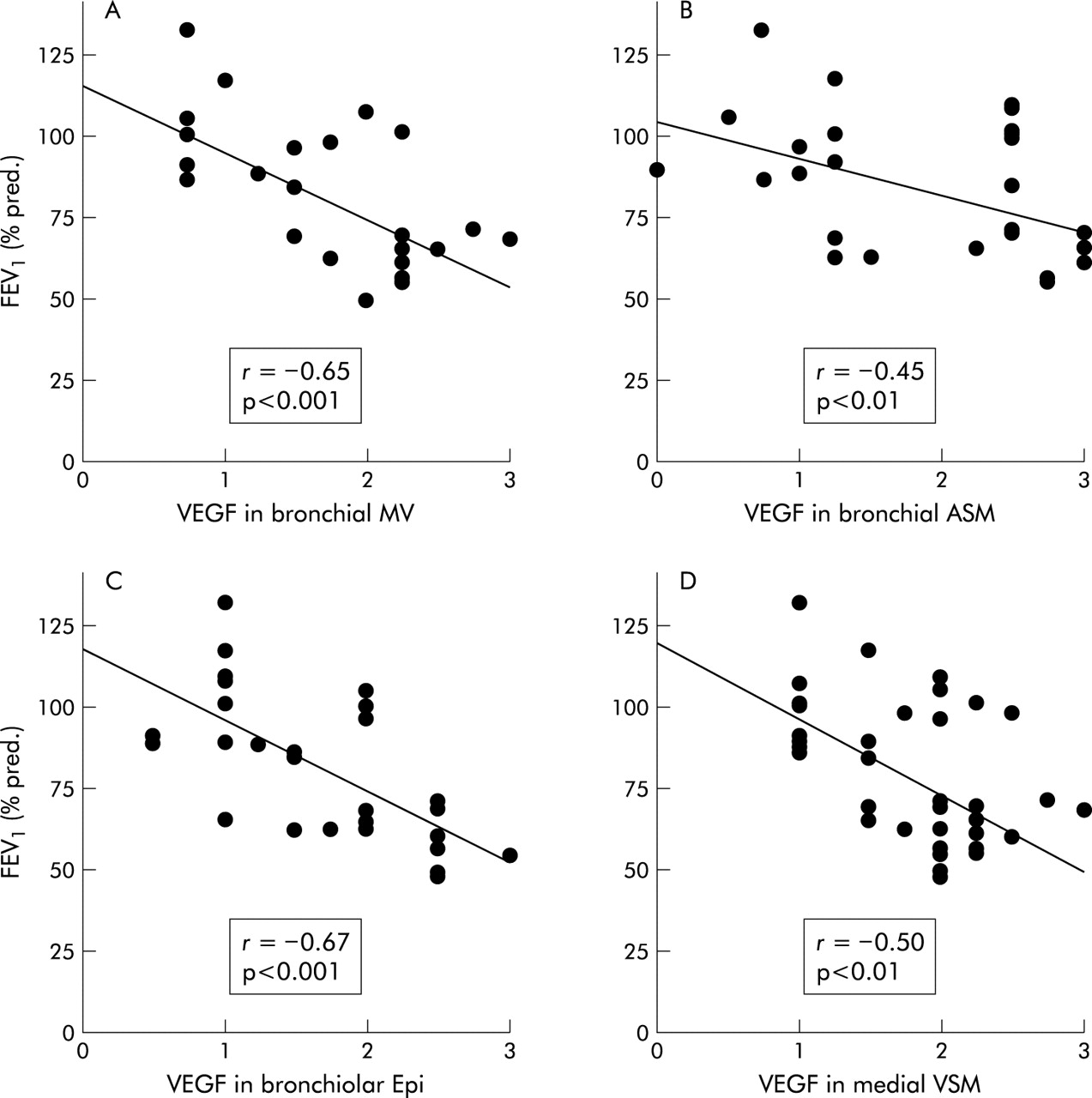
We examined the relationship between FEV1 values of patients in both groups and the staining scores of VEGF, Flt-1, and KDR/Flk-1 in the investigated areas. In the bronchial airways FEV1 values were inversely correlated with VEGF staining scores in the mucosal microvasculature (r = −0.65; p<0.001, fig 6A) and ASM cells (r = −0.45; p<0.01, fig 6B) if all subjects were analysed together. The VEGF staining scores in the bronchiolar epithelium (r = −0.67; p<0.001, fig 6C) and medial VSM cells of larger pulmonary arteries associated with the bronchiolar airways (r = −0.50; p<0.01, fig 6D) also showed an inverse correlation with FEV1 values from the total group. In addition, VEGF expression in the medial VSM cells was correlated with KDR/Flk-1 expression in the endothelium of the pulmonary arteries (r = 0.41; p<0.01) as well as the smaller alveolar vessels (r = 0.48; p<0.01). Furthermore, we found correlation for the expression pattern of KDR/Flk-1 and Flt-1 in the endothelium of the pulmonary arteries (r = 0.67; p<0.001) as well as in alveolar vessels (r = 0.80; p<0.0005).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlation with FEV1 (% predicted) of VEGF protein expression in (A) microvessels (MV) in the bronchial mucosa, (B) bronchial airway smooth muscle (ASM) cells, (C) bronchiolar epithelial (Epi) cells, and (D) medial vascular smooth muscle (VSM) cells of pulmonary arteries associated with the bronchiolar airways. Correlation was assessed for the combined patient groups (non-COPD and COPD). The correlation coefficient (r) was obtained using linear regression (Pearson’s) analysis.
We also examined the correlation between the epithelial VEGF expression from the current study and epithelial TGF-β1 expression from one of our previous studies.20 In the bronchiolar epithelium Pearson’s analysis revealed a significant positive correlation between the levels of VEGF protein and TGF-β1 protein (r = 0.55; p<0.004) and between VEGF protein levels and TGF-β1 mRNA expression (r = 0.45; p<0.02). In the alveolar epithelium VEGF protein levels correlated significantly with TGF-β1 mRNA expression only (r = 0.58; p<0.002), but not with TGF-β1 protein levels (r = 0.31; p<0.12).
DISCUSSION
The results of this study show that COPD is associated with increased expression of VEGF in the bronchial, bronchiolar, and alveolar epithelium and in bronchiolar macrophages as well as ASM and VSM cells in both the bronchiolar and alveolar regions. KDR/Flk-1 and Flt-1 were increased in patients with COPD compared with non-COPD subjects in endothelial, intimal and medial VSM cells of the larger pulmonary arteries and of smaller calibre alveolar vessels. Interestingly, we observed a significant inverse correlation between VEGF and FEV1 in bronchial mucosal microvessels and ASM cells, bronchiolar epithelium, and medial VSM cells of the larger pulmonary arteries associated with the bronchiolar airways. TGF-β1 staining in the bronchiolar epithelium also correlated with VEGF in the same patients as described in our previous study.20
Our results indicate that VEGF and its receptors Flt-1 and KDR/Flk-1 are localised within the airways and vasculature in endothelial and epithelial cells as well as smooth muscle cells and, furthermore, on various inflammatory cells (predominantly macrophages). The localisation of VEGF and its receptors in the lungs of our patient groups is in agreement with earlier reports which described a similar staining pattern in human developing lungs, normal adult lungs, and emphysematous lungs.17,27,28 In contrast to Kasahara et al28 who showed that VEGF and its receptor VEGF-R2 were decreased in total lung extracts of emphysematous lungs as measured by ELISA or western blot analysis, we found that the epithelial and endothelial cells in the alveolar spaces and in the most distal airways were intensely positive for VEGF and KDR/Flk-1 in patients with COPD. In our study the patients could be considered as having mild to moderate COPD whereas the lungs studied by Kasahara et al were solely emphysematous.
Our findings of increased VEGF expression in viable cell populations indicate in part a successful attempt to repair sustained damage, and perhaps a contribution to vascular remodelling and participation in the establishment and maintenance of the functional blood-gas interface, maturation, survival, and proliferation of capillary endothelial cells.29 In adult lungs VEGF and its receptor system may contribute to the maintenance of endothelial and epithelial cell viability in response to injury.31
Interestingly, immunoreactivity for VEGF in intimal and medial VSM cells and for Flt-1 and KDR/Flk-1 in endothelial cells of the pulmonary arteries and alveolar vessels was increased in patients with COPD. The highest levels of VEGF expression in the pulmonary vasculature were observed in the medial VSM cells and of KDR/Flk-1 in endothelial cells of arteries with a diameter of approximately 200 μm, which are known to play an important role in pulmonary blood pressure regulation and vascular resistance.14,30 Pulmonary hypoxia and hypertension with increased sheer stress are pathophysiological conditions that have been shown to increase the expression of VEGF in VSM cells.13,14 Blockade of KDR/Flk-1 is associated with obliterative endothelial cell proliferation in pre-capillary arterioles with abnormal vessel development and, at the same time, with induction of capillary endothelial and cell death by apoptosis, together leading to death in rat embryos similar to that seen in subjects with primary pulmonary hypertension.13,18,31,32 In a follow up study Tuder et al33 found that, after VEGFR-2 blockade, apoptosis predominated in areas of oxidative stress and apoptosis blockade by a broad spectrum caspase inhibitor markedly reduced the expression of markers of oxidative stress.33 Hypoxia, oxidative stress, and pulmonary hypertension are pathological features often seen in patients with advanced COPD, and increased VEGF expression may lead to increased or even abnormal proliferation of endothelial and VSM cells in pulmonary vessels. This suggests a potential role for this endothelial mitogen in peripheral angiogenesis and vascular remodelling, possibly in combination with other smooth muscle specific growth factors such as FGF-2, PDGF, and TGF-β1.12,34–36
We observed increased VEGF expression and unchanged Flt-1 and KDR/Flk-1 expression in bronchiolar and alveolar epithelial cells as well as in ASM cells in patients with COPD. It has previously been reported that the expression of VEGF and receptor KDR/Flk-1 can also be induced by stimuli such as hypoxia and oxidative stress in cells other than endothelial cells, such as epithelial and smooth muscle cells.33,37,38 In a recent report Kanazawa and colleagues39 showed that VEGF levels in induced sputum were higher in patients with bronchitis and lower in those with emphysema than in normal controls. Moreover, VEGF levels in patients with bronchitis were inversely correlated with FEV1 values. Our data on the inverse correlation of VEGF levels in various airway and vascular cells is in agreement with this report. In our study subjects with COPD could not be subdivided into patients with either chronic bronchitis or emphysema alone. Furthermore, the nature of the human material examined (sputum) in the study by Kanazawa and colleagues is different from the lung tissue in which we immunohistochemically localised and quantified VEGF and its receptors.
Recent studies have indicated that VEGF expression is increased in bronchial and alveolar epithelial cells and is also induced in α-SMA positive (myo)fibroblasts in bleomycin induced fibrosis in the rat and in human patients with pulmonary fibrosis, and that these fibrotic regions are densely populated by mast cells and macrophages with increased KDR/Flk-1 expression.15,17 We have shown previously that mast cells and macrophages are increased in bronchiolar airway epithelium and have reported increased expression of TGF-β1 in bronchiolar and alveolar epithelial cells in patients with COPD.20,21 We found a significant correlation between VEGF expression in epithelial cells and TGF-β1 expression published previously in the same patient groups,20 which suggests that the VEGF/Flk-1 system—possibly combined with TGF-β1—represents a molecular link between inflammatory cell accumulation and proliferation of myofibroblasts.
The increased expression of VEGF and TGF-β1 on bronchiolar epithelial cells and macrophages and the presence of KDR/Flk-1 and Flt-1 suggest a mechanism of initiating and perpetuating fibrosis at sites of tobacco induced injury contributing to airway remodelling in COPD. Inhaled corticosteroids may decrease the levels of VEGF expression,40 but this was not the case in our study as none of the patients had received inhaled corticosteroid treatment except four who received corticosteroids perioperatively. Caution must be exercised in extrapolating the expression data based on only 14 patients in each group, but the trend for increased VEGF expression in bronchial airways and KDR/Flk-1 in bronchial and bronchiolar airway smooth muscle could reach significance if more patients were included.
Taken together, these findings strongly suggest a role for VEGF and its receptors in airway and vascular remodelling, and thereby in the development of airway obstruction in COPD. At present our knowledge of airway and vascular remodelling during the development of COPD is far from complete. It is probable that many growth factors—including VEGF—play an essential role in the pulmonary and vascular viability and repair in response to tissue injury. Increased pulmonary VEGF expression in the airways, parenchymal lining, and small diameter pulmonary vessels in COPD may reflect a partly unsuccessful attempt to stimulate tissue repair mechanisms caused by tobacco induced injury.
Acknowledgments
The authors thank Drs J Stolk and J H J M van Krieken for their help in the analysis of the clinical data and pathology, respectively, and Mrs A Willems-Widyastuti for her technical assistance.
REFERENCES
Footnotes
-
This study was supported in part by The Netherlands Asthma Foundation (grants #97.73 and 95.49).
-
ARK and WIdB contributed equally to the work.
Linked Articles
- airwaves