Article Text

Abstract
Background: Tumour necrosis factor α (TNFα) is a major therapeutic target in a range of chronic inflammatory disorders characterised by a Th1 type immune response in which TNFα is generated in excess. By contrast, asthma is regarded as a Th2 type disorder, especially when associated with atopy. However, as asthma becomes more severe and chronic, it adopts additional characteristics including corticosteroid refractoriness and involvement of neutrophils suggestive of an altered inflammatory profile towards a Th1 type response, incriminating cytokines such as TNFα.
Methods: TNFα levels in bronchoalveolar lavage (BAL) fluid of 26 healthy controls, 42 subjects with mild asthma and 20 with severe asthma were measured by immunoassay, and TNFα gene expression was determined in endobronchial biopsy specimens from 14 patients with mild asthma and 14 with severe asthma. The cellular localisation of TNFα was assessed by immunohistochemistry. An open label uncontrolled clinical study was then undertaken in 17 subjects with severe asthma to evaluate the effect of 12 weeks of treatment with the soluble TNFα receptor-IgG1Fc fusion protein, etanercept.
Results: TNFα levels in BAL fluid, TNFα gene expression and TNFα immunoreative cells were increased in subjects with severe corticosteroid dependent asthma. Etanercept treatment was associated with improvement in asthma symptoms, lung function, and bronchial hyperresponsiveness.
Conclusions: These findings may be of clinical significance in identifying TNFα as a new therapeutic target in subjects with severe asthma. The effects of anti-TNF treatment now require confirmation in placebo controlled studies.
- BHR, bronchial hyperresponsiveness
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- PEF, peak expiratory flow
- TNFα, tumour necrosis factor α
- asthma
- tumour necrosis factor alpha (TNFα)
- bronchial hyperresponsiveness
- etanercept
- corticosteroid
Statistics from Altmetric.com
- BHR, bronchial hyperresponsiveness
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- PEF, peak expiratory flow
- TNFα, tumour necrosis factor α
Asthma is a disorder of the conducting airways characterised by Th2 mediated inflammation, with recruitment of a range of inflammatory cells and enhanced mediator release.1 In mild disease the inflammatory response, attendant bronchial hyperresponsiveness (BHR), and variable airflow obstruction are highly responsive to inhaled corticosteroids, positioning these drugs as first line controller therapy for this disease.2 However, in persistent and severe asthma, inhaled corticosteroids are only partially effective, and patients often require intermittent or continuous oral corticosteroids. This severe end of the disease spectrum, which exhibits an altered inflammatory cell profile involving neutrophils3,4 and accounts for approximately 10% of the asthmatic population, represents an important unmet clinical need as it is these patients who have the highest morbidity and mortality.5
Tumour necrosis factor α (TNFα) is a major therapeutic target in a range of chronic inflammatory disorders involving neutrophils. These include rheumatoid arthritis, juvenile arthritis, ankylosing spondylitis, Crohn’s disease, psoriasis, glomerulonephritis, sarcoidosis and Behcet’s disease,6 all of which are characterised by a Th1 type immune response associated with excess generation of TNFα. Although asthma is considered an eosinophilic disorder involving Th2 cytokines,1 there has been some interest in the TNF family of cytokines, especially TNFα. In murine models of asthma, a deficiency in TNFα receptors, chronic treatment with a TNFα antibody, or induction of a TNFα autoantibody results in marked attenuation of antigen induced airway inflammation.7,8 Genetic association studies have also shown a strong association between TNFα gene polymorphism and BHR9 and asthma,10 and inhalation of TNFα in both rodents11 and normal or asthmatic humans12,13 leads to the development of BHR accompanied by airway neutrophilia. Many different cell types produce TNFα, but those especially relevant to asthma include T lymphocytes, monocytes/macrophages, mast cells, eosinophils and epithelial cells.14 Importantly, TNFα promotes recruitment of neutrophils, as well as eosinophils, into the airways through functional effects on the endothelium15 and by direct and indirect chemotactic effects.16 Recognising the unmet need in severe persistent asthma, we investigated the potential of TNFα as a therapeutic target in this disease.
METHODS
The studies were approved by the Southampton and South West Hampshire local research ethics committees and volunteers gave their written informed consent.
Clinical assessment and subject classification
Healthy non-asthmatic subjects and mild asthmatic subjects were recruited from a research database of volunteers, and subjects with severe asthma were identified from outpatient clinics. Detailed clinical history and physical examination were performed on all subjects. Spirometric tests and assessment of asthma severity were in accordance with the BTS/SIGN guidelines.2 All subjects were tested for atopy by skin prick testing and were free from respiratory tract infections for a minimum of 4 weeks before inclusion in the study. The characteristics of the groups of subjects enrolled into the various arms of the study are summarised in table 1.
Subject details presented for the whole study group and for each separate study group
Fibreoptic bronchoscopy
Subjects underwent fibreoptic bronchoscopy under local anaesthesia as previously described17 using an Olympus BFIT20 bronchoscope (Olympus, Tokyo, Japan). Bronchoalveolar lavage (BAL) fluid was obtained by wedging the bronchoscope into a segmental bronchus and introducing six 20 ml aliquots of 0.9% saline, pre-warmed to 37°C. Gentle suction was used to collect the fluid into a 100 ml plastic trap. The lavage fluid was centrifuged at 400g for 10 minutes at 4°C and the supernatant aliquoted and stored at −80°C until assayed for TNFα. Bronchial biopsies were taken from the third or fourth airway carinae using Precisor Broncho pulmonary coated disposable biopsy forceps (Bard Endoscopic Technologies, Billerica, USA). These were either transferred immediately into ice cold acetone containing iodoacetamide (20 nM) and phenylmethylsulfonylfluoride (2 mM) and placed at −20°C for 24 hours before processing into glycol methacrylate resin (Polysciences, Northampton, UK) for embedding,18 or snap frozen in liquid nitrogen prior to RNA extraction.
Immunodetection of TNFα
BAL fluid was concentrated 10–30 fold using Amicon Centriprep YM-3 centrifugal filter devices (Millipore (UK) Ltd, Watford, UK) before measurement of TNFα by ELISA (R&D Systems, Abingdon, Bucks, UK). The assay had a lower limit of detection of 60 fg/ml and a coefficient of variation of repeated measures of 3.4%. Data are corrected for concentration and thus represent unconcentrated BAL fluid values.
Immunohistochemical staining for TNFα was conducted as previously described18,19 using a murine monoclonal antibody against human TNFα (1:500, R & D Systems). The specificity of the antibody was confirmed by omitting the primary antibody and replacing it with an isotype matched antibody. The number of TNFα positive cells was determined by counting the total number of TNFα immunoreactive cells within the biopsy section, excluding areas of muscle, large blood vessels, glands, damaged tissue and artefact. Counts were made in two separate tissue sections cut at least 10 μm apart, and the mean number of cells calculated. Only cells with an identifiable nucleus were counted. The area of the individual biopsy samples was measured using image analysis (Colorvision 1.7.6, Improvision, Coventry, UK), excluding those areas not included in the counting, and the data corrected to cells per mm2.
TNFα mRNA expression
For analysis of TNFα gene expression, total RNA was extracted from individual biopsy specimens. One microgram of total RNA was reverse transcribed using the reverse transcription (RT) System (Promega, Southampton, UK) according to the manufacturer’s instructions. TNFα primer (Qiagen) and probe (Eurogentec, Seraing, Belgium) sequences labelled with a 5′-reporter dye FAM (6-carboxy-fluorescin) and a 3′-Eclipse DARK quencher™ were: forward primer 5′-AAGAGGGAGAGAAGCAACTACAGA-3′; reverse primer 5′-GGTGGAGCCGTGGGTCAG-3′; and probe FAM-5′-AACAACCCTCAGACGCCACATCCCCT-3′-Eclipse. Random hexamers were used to reverse transcribe total RNA as 18S ribosomal RNA (18S rRNA) was the endogenous control. For each duplicate sample the PCR reaction contained 25 ng of cDNA template, 3.2 pM fluorogenic probe, 15 pM forward and reverse primers, and 7.5 µl qPCR mix (Eurogentec) in a final volume of 12.5 µl. Separate normalising controls were run which contained 1 µl of 18S rRNA and probe mixes (Eurogentec). RT negative samples were used to indicate that the signals obtained were RT dependent. The PCR protocol was as follows: 95°C, 10 minutes, followed by 40 cycles of denaturation 95°C, 15 seconds, and annealing/extension 60°C, 1 minute. Thermocycling and real time detection of PCR products were performed on an iCyclerIQ sequence detection system (Bio-Rad, Hercules, CA, USA) and, following completion of the PCR reaction, the thresholds for fluorescence emission baseline were set automatically at 10 times the background levels on the FAM layer. Expression levels were normalised to the 18S rRNA levels and then expressed relative to the lowest expression value found in mild asthma using the ΔΔCT method. Samples were measured in duplicate.
Proof of principle clinical study evaluating the effect of etanercept in severe asthma
Seventeen subjects (12 women) of median age 43 years (range 30–67) with a mean (SD) duration of asthma of 23.8 (11.7) years were enrolled into the trial. Their mean (SD) resting forced expiratory volume in 1 second (FEV1) was 68.3 (5.0)% predicted. These patients were similar to those studied in the bronchoalveolar lavage and biopsy studies, being at stage 5 of the BTS/SIGN asthma management guidelines,2 requiring treatment with high dose inhaled corticosteroids (equivalent to beclometasone 2500 μg/day) and oral prednisolone (mean dose 11.5 mg/day). In addition to long acting β2 agonists, theophylline and leukotriene modifying drugs, all the subjects required prn salbutamol on an as required basis, delivered either by metered dose inhaler or by nebuliser. Current smokers, subjects with a smoking history of more than 10 pack years, and those with other coexisting lung diseases, a history of tuberculosis, multiple sclerosis, lupus erythematosus, and other autoimmune diseases were excluded.
Each patient received subcutaneous etanercept (Enbrel, Wyeth Laboratories, Berkshire, UK) 25 mg twice weekly as add-on therapy for 12 weeks. The dose of medication and the duration of treatment were chosen from the initial trials of etanercept in rheumatoid arthritis.20 No change was made to their regular controller asthma medications during the 12 week treatment period and all reliever medications except bronchodilators were kept constant throughout the study.
The primary efficacy variable was improvement in asthma control and the secondary outcome measures were changes in airway hyperresponsiveness to inhaled methacholine and lung function. The manufacturer of etanercept had no involvement in the design of the study.
Subjective asthma control, as measured by the Juniper asthma control questionnaire,21 was recorded on entry and completion, together with morning and evening peak expiratory flow (PEF) using Wright mini peak flow meters (the best of three) recorded daily in diary cards. Adverse events during the study period and use of regular and rescue medication were also recorded. The clinic lung function was recorded as FEV1 and forced vital capacity (FVC) using a Vitalograph compact spirometer (Vitalograph Ltd, Maids Moreton, Bucks, UK). Spirometric tests were performed at baseline, on entry into the study, and at the end of the study as well as at a follow up visit 8 weeks after the last dose of the study drug.
Non-specific BHR was assessed before and at the end of week 12 of treatment with etanercept using the methacholine bronchial provocation test.22 The test was carried out in all subjects whose FEV1 was more than 50% predicted. Subjects were asked not to take their salbutamol (or other rescue bronchodilators) within at least 4 hours before the methacholine challenge. Nebulised methacholine (Sigma Co, Poole, Dorset, UK) dissolved in saline was administered through a dosimeter (Spira Electro 2, Spira, Finland) in doubling dilutions until the FEV1 fell by at least 20%. The degree of airways responsiveness to methacholine was expressed as the cumulative PC20, as determined by linear interpolation.22
Sputum induction was performed on all the subjects at baseline and after 12 weeks of treatment with etanercept according to the method of Pizzichini et al23 using inhalation of aerosolised hypertonic saline (4.5%) for 20 minutes administered via an ultrasonic nebuliser (Devilbiss Ultraneb 2000, PA, USA). The procedure was discontinued when there was a fall in PEF of >15% or if there were troublesome symptoms. Sputum processing involved adding an equal weight of 0.01M dithioerythritol, filtering through a 70 µm filter, and centrifugation for 10 minutes at 400g at 4°C. The cell pellet was resuspended in 1 ml Tris-buffered saline (TBS) and cells were counted in a Neubauer’s chamber after staining with trypan blue. Two cytospins per sample were processed using Rapi-Diff stain and 600 cells counted per cytospin, in a blinded fashion, to obtain differential cell counts; the mean value was used for analysis. The differential cell counts were expressed as the percentage of the total cells.
Statistical analysis
Paired t test (mean (SE)) and Wilcoxon’s signed rank tests (median, IQR) were used as appropriate for within group comparisons and non-paired comparator analyses with t tests and Mann-Whitney U tests were used for between group comparisons. Statistical analysis was undertaken using SPSS for Windows version 11.5 (SPSS, Chicago, IL, USA).
RESULTS
TNFα levels in BAL fluid and mRNA
As the airway lumen is an important site of neutrophil accumulation in severe asthma,3,4 we first sought evidence for abnormal levels of TNFα in BAL fluid from asthmatic subjects with varying degrees of disease severity (table 1) and non-asthmatic controls. The severe asthmatics had significantly higher concentrations of TNFα (median 160 fg/ml (range 100–6660)) in BAL fluid than either the healthy controls (117 fg/ml (64–301)p = 0.001) or those with mild asthma (111 fg/ml (37–322), p<0.001); the difference between the latter two groups was not significantly different (fig 1A). Relative TNFα mRNA levels were also significantly higher in biopsy specimens from the severe asthmatic subjects (median 2617 (range 58–276 488)) compared with mild asthmatic subjects (84 (1–1282); p = 0.002, fig 1B).
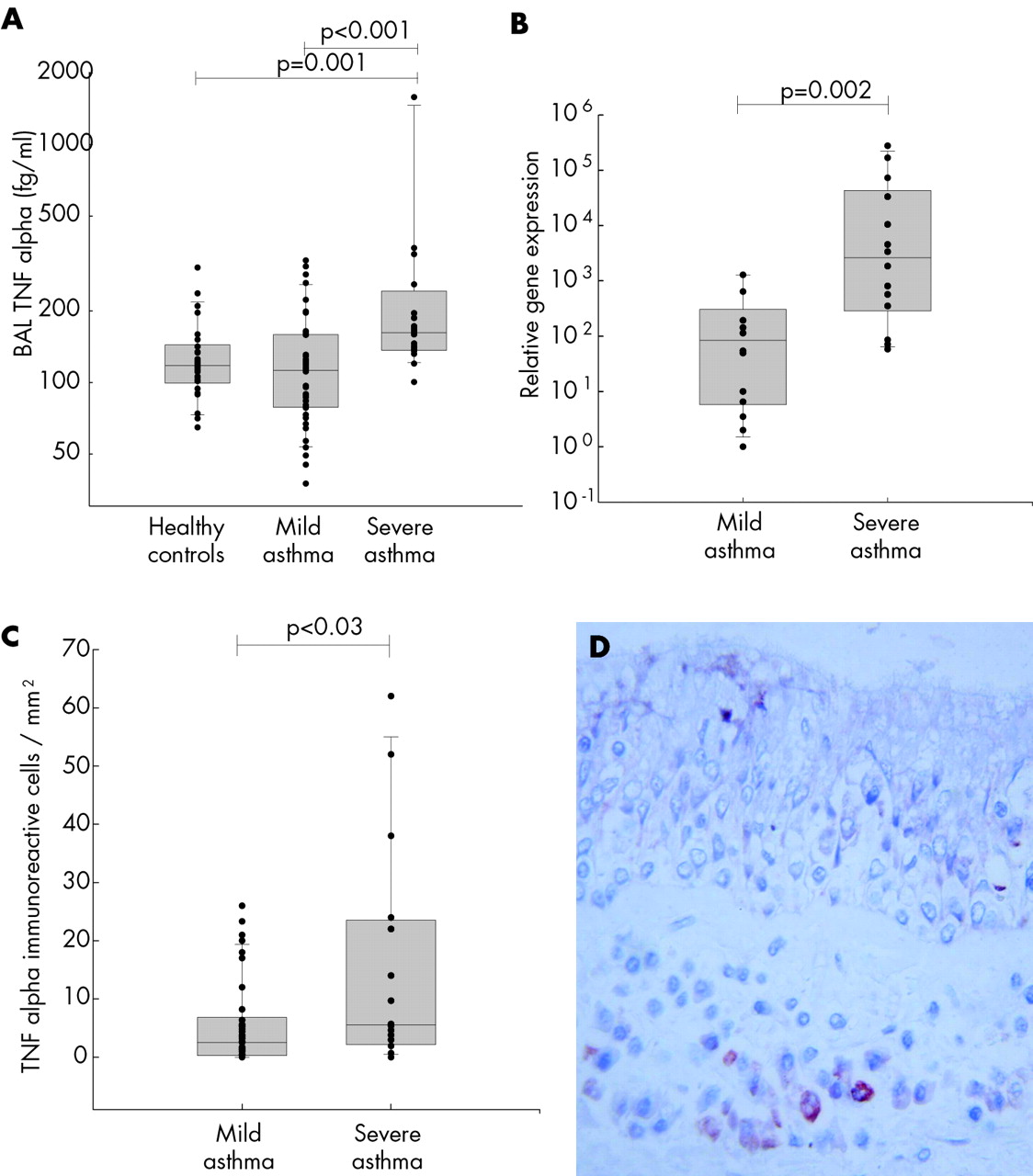
(A) TNFα concentrations in bronchoalveolar lavage (BAL) fluid from healthy control subjects (n = 26), subjects with non-steroid treated mild asthma (n = 42) and those with steroid treated severe asthma (n = 20). For presentation purposes, one outlier (6660 fg/ml) in the severe asthma group is omitted from the graph. TNFα was measured by ELISA. (B) TNFα gene expression in endobronchial biopsy specimens from 14 patients with mild asthma and 14 with severe asthma. Total RNA was extracted and analysed by reverse transcription quantitative PCR. Results are normalised to the 18S rRNA levels and are presented as relative TNFα levels which were calculated using the ΔΔCT method. (C) Immunoreactive TNFα positive cell counts in endobronchial biopsy specimens (cells/mm2) from 42 subjects with mild asthma and 14 subjects with severe asthma. (D) Immunohistochemical staining and localisation of TNFα in submucosal cells in a bronchial biopsy specimen from a subject with severe asthma. The data in A, B and C are presented as box and whisker plots showing the median, interquartile range, and 95% confidence limits with individual data points superimposed. Data were analysed using the Mann-Whitney U test and significant differences are indicated.
TNFα levels in bronchial biopsy specimens
To further identify the cellular source of TNFα protein, tissue sections of bronchial biopsies were examined by immunohistochemistry. This showed that TNFα expression was localised predominantly to mast cells, with occasional other cell types also showing some positive immunostaining. There were significantly greater numbers of TNFα immunoreactive cells/mm2 in biopsy specimens from subjects with severe asthma (median 5.6 (range 0–62)) than in those from subjects with mild asthma (median 2.5 (range 0–26); p<0.03, fig 1C and D).
Clinical study with etanercept
Seventeen patients (table 1) with severe asthma who were symptomatic despite receiving maximum inhaled corticosteroids were recruited into the open label study and received etanercept as add-on therapy for 12 weeks. Two of the 17 subjects enrolled into the study failed to complete it, one because of a swelling in the neck that was subsequently identified as a lipoma and the second developed a skin rash with the first dose of etanercept and was not keen to continue in the trial despite the rash resolving spontaneously. The adverse events during etanercept treatment were mild (table 2).
Adverse events during treatment with etanercept
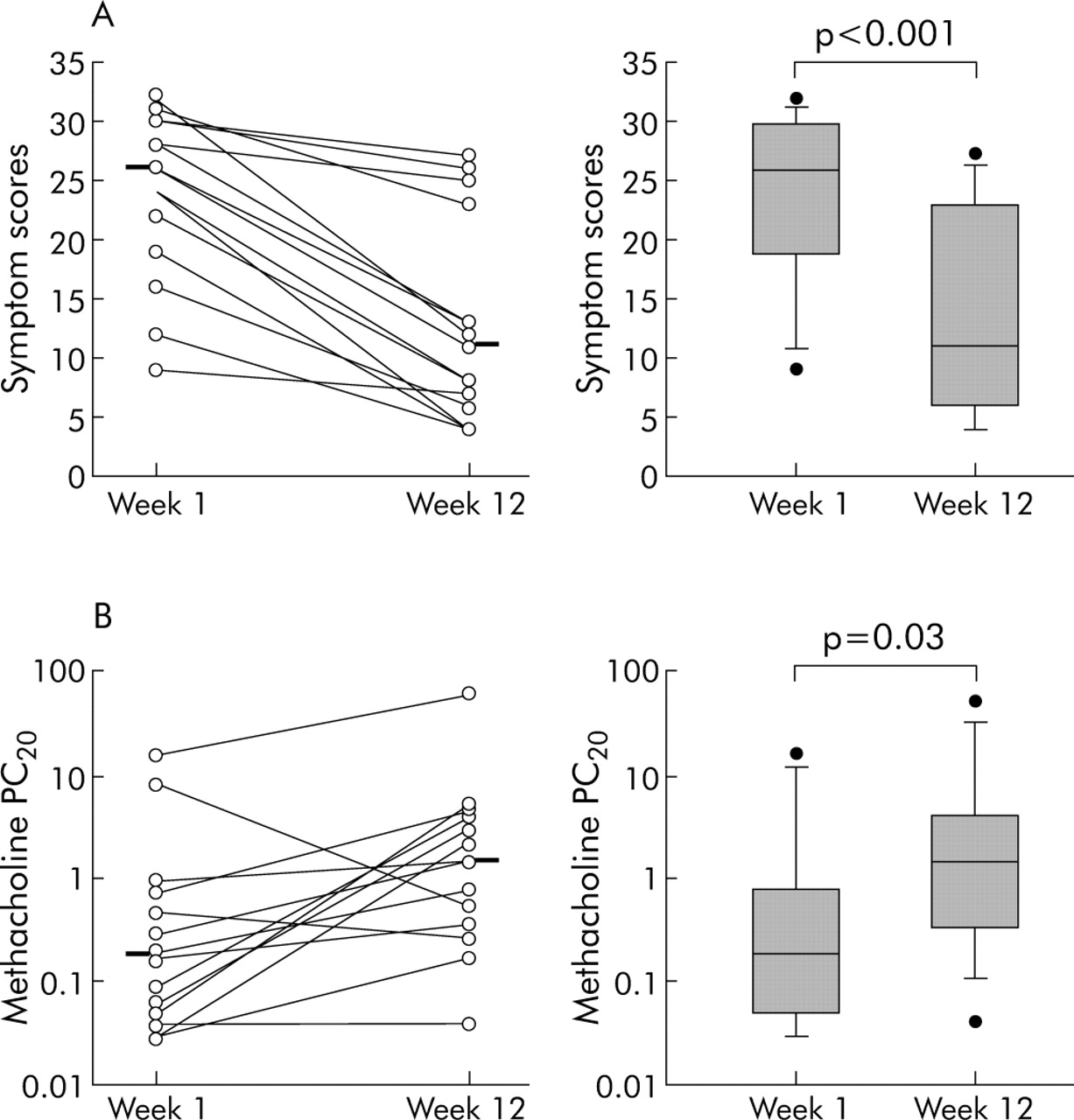
Of the 15 subjects who completed the study, 12 weeks of treatment with etanercept resulted in a marked and highly significant improvement in symptoms with the mean (range) asthma control questionnaire score falling from 26 (9–32) to 11 (4–27), p<0.001 (fig 2A). This was accompanied by a significant increase in baseline FEV1, FVC, and morning and evening PEF (table 3), and all but one of the patients discontinued their use of nebulised β2 agonist. Of particular significance was the effect of etanercept in reducing BHR, with the provocative concentration of methacholine producing a 20% decrease in FEV1 (PC20) increasing from a geometric mean of 0.21 (0.1–0.64) mg/ml to 1.28 (0.53–2.92) mg/ml (fig 2B), representing a change of 2.5 doubling dilutions (table 3). Eight weeks after stopping etanercept, symptom scores and lung function had returned to pretreatment values.
Changes in lung function before and after treatment with etanercept

{kind=link}
{kind=link}
(A) Symptom scores before and after treatment in 15 corticosteroid dependent severe asthmatic subjects treated with etanercept 25 mg twice weekly for 12 weeks. (B) Concentration of methacholine required to reduce FEV1 by 20% of baseline (PC20) at weeks 1 and 12 as a measure of the effect of etanercept on bronchial reactivity. Data are shown as paired data before and after treatment with median (panel A) or geometric mean (panel B) indicated (left hand panels); the box and whisker plots (right hand panel) show median (panel A) or geometric mean (panel B), interquartile range and 5–95% confidence intervals with outliers shown as dots. Statistical significances were determined using the Wilcoxon rank sum test.
Despite the severity of their asthma, we were able to obtain paired sputum samples from 11 of the 15 subjects involved in the study. However, although reductions in eosinophil and neutrophil numbers were observed in eight of the 11 subjects, this failed to achieve statistical significance.
DISCUSSION
Although asthma is considered an eosinophilic disorder,1 at the severe end of the disease spectrum there is an altered inflammatory cell profile involving neutrophils.3,4 Consistent with this predominance of neutrophils,3,4 we have shown that patients with severe corticosteroid dependent asthma have higher concentrations of TNFα in recovered BAL fluid. In contrast, no difference was seen between BAL fluid TNFα concentrations in mild asthmatics and healthy controls. This suggests that the increase in TNFα seen in subjects with severe disease is a feature of more persistent and corticosteroid refractory asthma rather than of asthma per se. This extends previous reports in which increased TNFα concentrations have been reported in BAL fluid in symptomatic asthma24 and in those ventilated because of acute severe disease.25 We also found greater numbers of TNFα immuno-reactive positive cells and greater gene expression for TNFα in endobronchial biopsy specimens from subjects with severe asthma than from those with symptomatic non-steroid treated asthma. As we have previously reported in biopsies from mild asthma,19 this TNFα was localised predominantly to mast cell granules. Our findings in severe asthma were evident despite treatment with high dose inhaled corticosteroids and, in the majority, with additional oral corticosteroid therapy. Thus, not only is TNFα overexpressed within the airways of patients with severe disease, but current optimal treatment does not resolve this.
The therapeutic options for patients with corticosteroid dependent asthma are limited. These patients represent a clinical burden accounting for approximately 30% of the healthcare costs of asthma through multiple hospital admissions for exacerbations and the side effects of long term corticosteroids.5 In other disease areas the appreciation that anti-TNFα strategies are able to modify disease that persists despite corticosteroid therapy led us to consider the potential benefit of anti-TNFα intervention strategies in the severe asthmatic group. We opted to use etanercept, which binds specifically to both TNFα and TNFβ thereby preventing free cytokine binding to cell surface TNF receptors.26 This was a proof of concept study and, as such, was open labelled and uncontrolled.
Etanercept treatment was associated with improvement in asthma symptoms, lung function, and BHR. It is unlikely that such changes would have arisen by chance in view of the chronic and persistent nature of the disease in these patients. However, the study cannot be considered conclusive as it was an open labelled study. Despite maximum doses of inhaled and oral corticosteroids, regular treatment with etanercept produced remarkable improvements in both clinical and physiological measures of asthma. The most striking feature was the improvement in BHR. When measuring BHR in asthma using repeated methacholine challenge, it is accepted that the error in PC20 determination is within one doubling dilution. Following experimental allergen exposure or natural allergen exposure, changes of 1–2 doubling dilutions in PC20 are considered clinically significant.27 Thus, although we had no data on which to base a power calculation before starting the study, our findings of an improvement of 2.5 doubling dilutions in PC20 is well outside the natural variation and consistent with a significant clinical improvement, in addition to any beneficial effect that may have already been achieved using inhaled and oral corticosteroids. At the end of the study the beneficial effects of etanercept were maintained for 2–4 weeks before asthma symptoms gradually returned to the pretreatment state.
The most common adverse effects encountered with etanercept were injection site reactions. The respiratory tract infections experienced by the four subjects would not be unusual in such patients with severe disease. It is difficult to suggest a causal association between the respiratory tract infections and the study medication as this was an open labelled study. The infections were associated with worsening of asthma control which was managed by increasing the dose of rescue medications. However, none of the subjects needed an increase in either their oral or inhaled corticosteroids, suggesting a possible beneficial effect of etanercept in these subjects. Controlled studies have shown that only 2% of patients with rheumatoid arthritis became antibody positive when treated with etanercept,28 so we did not evaluate the occurrence of antibodies in our study group. However, since immunogenicity appears to be associated with an increased incidence of infusion reactions and a shortened duration of clinical response, this needs to be investigated in larger studies.
An important target for TNFα is the microvascular endothelium with upregulation of adhesion molecules such as ICAM-1 and VCAM-1 and enhancing leucocyte migration and activation.15 We attempted to study the effects of etanercept on indices of inflammation using induced sputum. Although reductions in eosinophil and neutrophil numbers were observed in eight of the 11 subjects, this failed to achieve statistical significance. While this may have arisen by chance, our data raise the possibility that the beneficial effects of etanercept were unrelated to inhibition of inflammatory cell recruitment. However, the observation that inhaled TNFα increases BHR and leucocyte influx into human airways12,13 suggests that further studies involving tissue biopsies before and after therapeutic intervention may better address the effects of etanercept on endothelial cell activation and inflammatory cell recruitment.
Although etanercept and the anti-TNFα antibody infliximab both show powerful TNFα neutralisation, only infliximab is able to bind lamina propria T cells and induce apoptosis of activated lymphocytes in Crohn’s disease.29 We therefore believe it is unlikely that the beneficial effect of entanercept in severe asthma involves regulation of T cells. Since we found that the predominant cells that expressed TNFα in the airway biopsies were mast cells, at least some of the beneficial effects of etanercept are likely to relate to mast cell function. These cells selectively accumulate within the airways smooth muscle in asthma30 where the local release of TNFα can affect smooth muscle reactivity by increasing transcription of Gαi.31 As mast cell expression of TNFα is insensitive to inhibition by corticosteroids,32 its effect on smooth muscle function to cause BHR13 may be critical in maintaining persistent disease expression in chronic asthma. The modification of BHR by entanercept is therefore likely to be important in contributing to the clinical improvement. BHR is a fundamental physiological abnormality in asthma and, as corticosteroid insensitive BHR is a characteristic feature of severe disease,4,33 identification of a therapeutic intervention that reduces this is of major significance.
In summary, our study provides further evidence for a role for TNFα in severe asthma and is the first study to evaluate the effects of TNF blockade in patients with severe asthma. While TNFα may well play a part in the less severe forms of asthma associated with allergen exposure, the dominance of Th2 mediated effects in this subtype of disease suggests that TNFα blockade may not be so effective. There is now a need for large placebo controlled trials using both the soluble receptor and antibody approaches to blocking TNFα with a focus on severe corticosteroid dependent asthma, where there remains a large unmet clinical need.
Acknowledgments
The authors thank Miss Lorraine Hewitt for providing nursing care in association with bronchoscopies and Dr Rob M Powell who provided probe and primer sequences for TNFα quantitative PCR.
REFERENCES
Footnotes
-
Published Online First 15 September 2005
-
This work was supported by the Medical Research Council (UK) and an educational grant from Immunex (Amgen).
-
Competing interests: The proof of principle trial with etanercept was an investigator initiated study that was designed, executed, and analysed without input from any pharmaceutical company. Immunex (Amgen) provided an educational grant that contributed to the costs and Wyeth provided the etanercept without charge. The authors have no other competing interests to declare.
-
PHH and KSB contributed equally to this work.
Linked Articles
- Airwaves