Article Text

Abstract
The biochemical and clinical efficacy of intravenous augmentation therapy in α1-antitrypsin deficiency is reviewed, adverse events experienced with this treatment are considered, and its cost effectiveness is discussed.
- α1-antitrypsin deficiency
- augmentation therapy
Statistics from Altmetric.com
Because unopposed elastolysis is thought to be the mechanism by which emphysema develops in individuals with severe deficiency of α1-antitrypsin (AAT), the mainstay of current specific treatment for AAT deficiency is intravenous administration of purified AAT, so-called intravenous augmentation therapy. Pooled human plasma AAT has been licensed for prescription in several countries including the US, Canada, Italy, and Spain. This paper reviews the rationale for intravenous augmentation therapy and current evidence regarding its biochemical and clinical efficacy. After reviewing the specific criteria by which the biochemical and clinical efficacy of augmentation therapy can be judged, we then consider the available relevant studies that address efficacy. We also review the available data on adverse events experienced with augmentation therapy and its cost effectiveness, again by analysing the methodological features of available reports.
CRITERIA FOR DEMONSTRATING EFFICACY OF AUGMENTATION THERAPY
Criteria that must be satisfied in order to deem intravenous augmentation therapy effective should consider both the biochemical and clinical effects.1 Those for biochemical efficacy include:
-
evidence that intravenous augmentation therapy raises serum levels above the protective threshold and does so over the entire interdose interval;
-
evidence that the functional capacity of infused pooled human plasma AAT to oppose neutrophil elastase is preserved after the drug is infused.
Beyond these biochemical considerations, criteria that must be satisfied to assure clinical efficacy include:
-
evidence that the intravenous infusion of augmentation therapy slows the progression of emphysema or confers other clinical benefits such as decreased morbidity or enhanced survival;
-
evidence that augmentation therapy can be administered safely.
In considering the available evidence regarding the biochemical efficacy of intravenous augmentation therapy, Wewers et al2 first showed that the intravenous infusion of pooled human plasma AAT at a dose of 60 mg/kg once weekly produced serum levels that generally exceeded the protective threshold over the full dosing interval. As shown in fig 1, serum levels in 21 PI*ZZ recipients rose quite steeply acutely after infusion (to >300 mg/dl) and pre-dose nadir levels generally exceeded 11 μM (or 80 mg/dl), a level deemed to be the protective threshold above which the risk of emphysema is felt to be minimal but below which the risk rises. Indeed, nadir serum levels of AAT tended to rise over serial infusions, militating against the development of blocking antibodies. Furthermore, infused AAT maintained its functional anti-elastase activity, both in the serum and in the epithelial lining fluid (ELF).

Serum levels of α1-antitrypsin 30 minutes, 2 days, 4 days and 7 days after intravenous administration of 60 mg/kg body weight α1-antitrypsin. Each symbol represents the serum level for an individual subject receiving augmentation therapy. Reproduced from Wewers et al2 with permission of the publisher.
Subsequent studies have examined the biochemical efficacy of intravenous augmentation therapy administered at intervals other than weekly.3,4 Barker et al3 examined the pharmacokinetics of bi-weekly infusions of pooled human plasma AAT at a dose of 120 mg/kg in 23 PI*ZZ recipients who received 10 infusions each. In contrast to the pharmacokinetics of weekly augmentation therapy, serum levels above the protective threshold level of 80 mg/dl were achieved but were not sustained over the entire 2 week dosing interval. In no patient did the level remain >80 mg/dl over the full 14 days. In 41% (9/22) the levels were above 80 mg/dl for 7 days. Bronchoalveolar lavage was performed in five patients and epithelial lining fluid levels correlated poorly with serum levels (r = 0.60). Forced expiratory volume in 1 second (FEV1) was unchanged (1.22 l) among the recipients over a 20 week follow up period.3
Given the preference of both clinicians and patients for less frequent dosing of intravenous medications, the administration of monthly augmentation therapy has also been examined. Hubbard et al4 administered 250 mg/kg purified AAT intravenously once a month for 12 months to nine subjects with severe AAT deficiency (serum AAT levels <35 mg/dl or approximately 5 μM). As with weekly infusions, the nadir serum levels rose gradually over time, so that by the 12th month serum levels exceeded 80 mg/dl for a mean of 25 days of the 28 day dosing interval. Serum antineutrophil elastase activity was preserved and also rose over time, and ELF levels of AAT averaged 2.5 μM at day 28, thereby exceeding the theoretical ELF protective threshold value. No significant change in lung function was observed in recipients over the 12 month study period.4
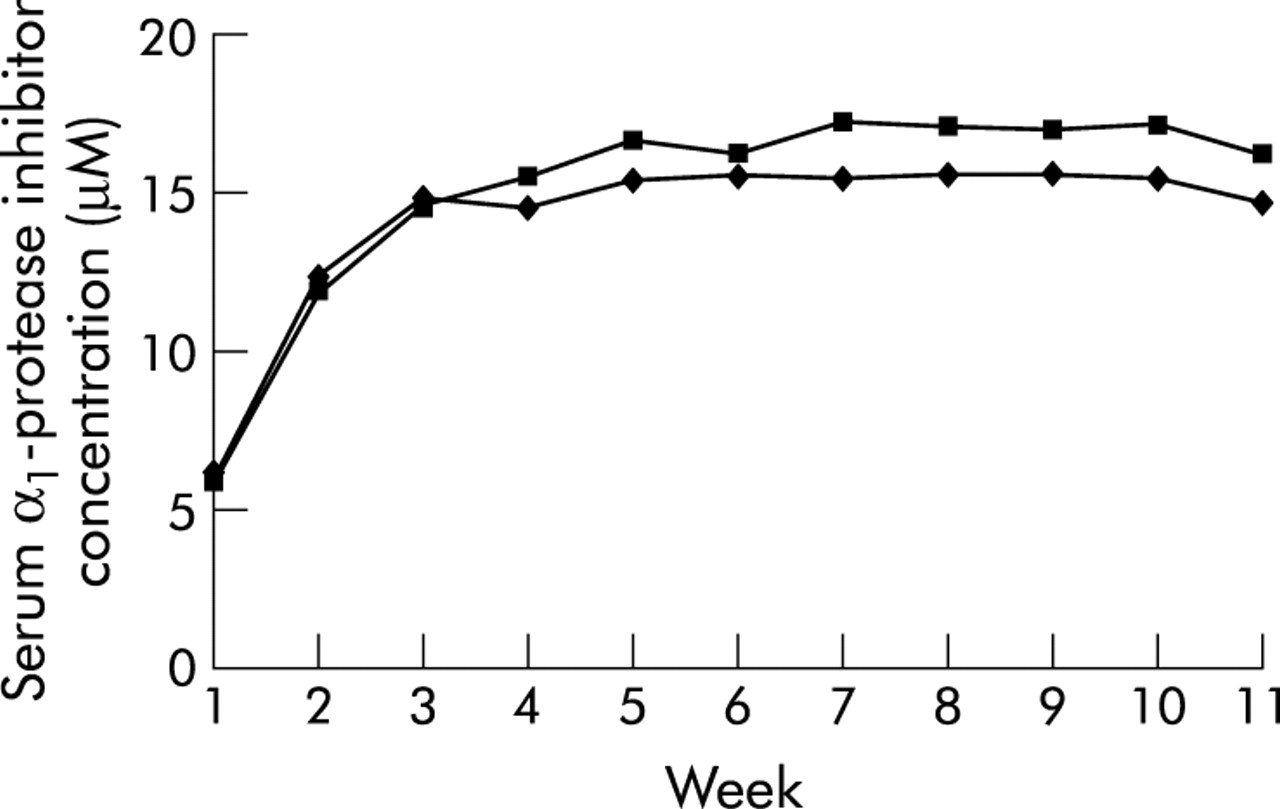
Since these early studies of the first available preparation (pooled human plasma AAT prepared by pasteurisation (Prolastin, Bayer, West Haven, CT, USA)), more recent studies have examined the biochemical profile of a different pooled human plasma AAT preparation that is purified using solvent detergent and nanofiltration techniques (Aralast, Baxter, Westlake Village, CA, USA).5 Using Prolastin as a comparator, the newer drug was evaluated in a four centre, randomised, double blind, controlled trial in which 13 subjects received each of the preparations for 11 weekly infusions (of 60 mg/kg), after which an open label follow up study with the new drug was conducted. As shown in fig 2, administration of this newer preparation satisfied equivalence criteria to the earlier approved preparation. Trough serum AAT levels at weeks 8–11 were similar with the new preparation (ratio 0.905 to comparator) and the slope of trough levels from weeks 12 to 24 did not exceed −0.1. No serious adverse events associated with the drug were observed. On the basis of this study, the new preparation received approval by the United States Food and Drug Administration (FDA) in December 2002.6 A third preparation (Zemaira, ZLB Behring) has since received US FDA approval.
{kind=link}
{kind=link}
Serum α1-protease inhibitor levels following intravenous augmentation with 60 mg/kg active Aralast (♦; n = 13) or controls (▪; n = 13). The number of weeks of augmentation therapy with either Aralast or control α1-protease inhibitor is shown on the abscissa. The slope of the curve is in μM/l/day. Reproduced from Stoller et al5 with permission of the publisher.
EVIDENCE FOR CLINICAL EFFICACY OF INTRAVENOUS AUGMENTATION THERAPY
Available studies of the clinical efficacy of augmentation therapy describe a variety of study designs,5,7–10 including observational studies (some with concurrent controls) and a single randomised, controlled clinical trial.8
Outcome measures in the available clinical studies include the rate of FEV1 decline, mortality rate, rate of change of lung density as measured by CT scanning, and the frequency of acute exacerbations and/or infection.11 Other clinical markers include inflammatory markers of sputum and ELF, as well as elastin breakdown products in urine.12
Table 1 presents a summary of the seven major available studies of the clinical efficacy of augmentation therapy.7–13 The largest studies are observational cohort studies with concurrent control groups. In the earlier of these two studies, Seersholm et al9 compared the rate of FEV1 decline in two patient groups—a treatment group of 198 PI*ZZ ex-smoking German individuals receiving Prolastin at 60 mg/kg weekly for at least 1 year and a control group of 97 Danish ex-smokers with severe AAT deficiency (PI*ZZ or ZNull) who were not receiving augmentation therapy. Baseline comparison of the groups showed overall similarity, with mild differences reflecting a slightly lower FEV1 among the treated group (mean 37% v 42% predicted, p = 0.02), a slight excess of men in the treated group (72% v 57%, p = 0.01), and a slightly shorter mean follow up duration in the treated group (3.2 v 5.8 years, p<0.01). Comparison of the rate of FEV1 decline showed that, overall, the rate was lower in those who received augmentation therapy than in the untreated group (mean −53 ml/year v −75 ml/year, p = 0.02). In a subset analysis by FEV1 strata the overall group difference was attributable to the slower decline in those whose FEV1 was 31–65% predicted, in which the treated group had a rate of FEV1 decline of −62 ml/year compared with −83 ml/year in the untreated group (p = 0.04).
Studies on clinical efficacy of augmentation therapy
The largest available observational cohort study was reported 1 year later in the National Heart, Lung, and Blood Institute (NHLBI) Registry for Individuals with Severe Deficiency of AAT.7 In this observational cohort study of 1129 individuals, 747 of whom received augmentation therapy at some time during Registry follow up, primary outcome measures were the rate of change in FEV1 and the mortality rate. For the overall group the mean rate of FEV1 decline was −54 ml/year. Features associated with an accelerated rate included: male sex, age 30–44 years, current smoking status, FEV1 39–79% predicted, bronchodilator responsiveness (defined as a rise in 12% and 200 ml of FEV1 after inhaled bronchodilator), and non-use of augmentation therapy. Although overall there was no significant difference in the rate of FEV1 decline between individuals receiving and those not receiving augmentation therapy, subset analysis showed a slower rate of decline in FEV1 among those who received augmentation therapy with American Thoracic Society stage II COPD (FEV1 35–49% predicted). Specifically, the decline in FEV1 in individuals who did not receive augmentation therapy was 27 ml per year faster than in those who did (p = 0.03).7 In addition to the rate of FEV1 decline, the mortality rate in subjects receiving and not receiving augmentation therapy was also considered in this study.7 Adjusting for other predictors, the risk ratio for death in augmentation therapy recipients was 0.64, significantly lower than in non-recipients (p = 0.02). In the subset of patients with stage II COPD, the risk ratio for death in those receiving augmentation therapy was 0.21 (p<0.001).7
Taken together, the concordant results from these two observational studies with concurrent controls suggest that augmentation therapy has clinical efficacy with a slowing in the rate of decline in lung function, particularly in individuals with moderate chronic airflow obstruction. However, as pointed out in the analysis of these study results, neither is a randomised trial so differences in the outcomes of compared groups could reflect differences between the groups for which statistical modelling, however sophisticated, could not adjust.
In addition to these two observational studies, Wencker et al studied the rate of decline in FEV1 before and after starting augmentation therapy in 96 patients with severe AAT deficiency (all with serum level <35% normal) and moderate airflow obstruction (mean FEV1 41% predicted).10 The overall rate of FEV1 decline was greater before initiating augmentation therapy than after (mean −49 ml/year v −34 ml/year, p = 0.019). Subset analysis showed that the effect of augmentation therapy achieved statistical significance in patients whose baseline FEV1 exceeded 65% predicted (mean −49 ml/year v −123 ml/year, p = 0.045) and in those who experienced a rapid decline in FEV1 (from above 65% predicted) before beginning augmentation therapy (p = 0.001). In the subset of patients with moderate airflow obstruction at baseline (FEV1 30–65% predicted) a trend was observed towards a slower rate of FEV1 decline after initiating augmentation therapy (p = 0.066).10
Other observational studies that have addressed the clinical efficacy of augmentation therapy include a web based survey of the frequency of infectious episodes by Lieberman,11 a study of urine desmosine levels after beginning augmentation therapy by Gottlieb et al,12 and an observational study of sputum inflammatory markers by Stockley et al.13 Lieberman11 posted a questionnaire on the web to a users’ group of approximately 300 PI*ZZ individuals of whom 48% responded; 96 individuals had received augmentation therapy and 47 had not. The frequency of lung infections per year reported in those who had used augmentation therapy was greater before the start of treatment than after treatment was initiated (65% v 18% reported experiencing >2 infections per year, p<0.001). Also, among non-recipients of augmentation therapy 55% reported experiencing >2 infections per year (p<0.001). Although this study has methodological shortcomings (possible selection bias, self-reported data), its results are consistent with the hypothesis that augmentation therapy reduces the rate of lung infections in PI*ZZ individuals.
Using urine desmosine as a measure of elastin breakdown, Gottlieb et al12 reported serial desmosine levels in 12 patients with severe AAT deficiency before and for 8 weeks after receiving augmentation therapy. In contrast to earlier results in two patients,14 urinary desmosine levels did not decline after augmentation therapy started, a result which may reflect the difficulty of using desmosine as a specific marker of lung elastin.
In the most recent observational study of clinical efficacy, Stockley et al13 analysed sputum and serum AAT levels and serum markers of inflammation in 12 PI*Z individuals given intravenous Prolastin at 60 mg/kg/week for 4 weeks. Affirming the biochemical efficacy first shown by Wewers et al,2 nadir serum AAT levels rose and were above the protective threshold value at week 4 (mean 14.4 μM). The rise in sputum AAT levels was accompanied by a significant increase in elastase inhibitory capacity and a significant decline in sputum leukotriene B4 levels, supporting the biochemical efficacy of intravenous augmentation therapy in the airway and its anti-inflammatory role.
Complementing these six observational studies of intravenous augmentation therapy, a single randomised controlled trial has been performed. Dirksen et al8 randomly allocated 28 PI*ZZ subjects to receive AAT infusions intravenously (250 mg/kg once monthly) for at least 3 years and another 28 PI*ZZ subjects to receive a placebo infusion (of human albumin). No difference was seen in the rate of decline in FEV1 between the two groups using either self-performed daily spirometry results or laboratory based spirometry results performed every 3 months (−59 ml/year v −79 ml/year, p = 0.25). However, when CT densitometry results were considered (in which slices 5 cm below the main carina were analysed using a 15th percentile threshold criterion for determining emphysematous lung), a trend was observed towards a slower annual rate of loss of lung density in augmentation therapy recipients (by 1.07 g/l/year). This difference approached but did not achieve statistical significance (p = 0.07).
Taken together, these studies demonstrate the biochemical efficacy of intravenous augmentation therapy, especially when given weekly. Clinical efficacy is suggested by the concordant results of several observational studies showing that augmentation therapy recipients experience a slower rate of decline in FEV1 and fewer lung infections, enhanced survival, and reduced lung inflammation as measured by sputum markers. Also, the results of the single randomised, placebo controlled trial show a trend towards a slower loss of lung density in augmentation therapy recipients.
ADVERSE EFFECTS EXPERIENCED WITH INTRAVENOUS AUGMENTATION THERAPY
Experience with intravenous augmentation therapy in several thousand American recipients for more than 14 years suggests that the treatment is generally well tolerated.
As shown in table 2, two large studies have specifically addressed the frequency and types of adverse effects associated with intravenous augmentation therapy. Wencker et al15 described the experience of 443 Prolastin recipients, of whom 65 experienced 124 adverse events. Five of these reactions were deemed severe and required medical intervention or hospitalisation (four anaphylactic reactions and one case of congestive heart and respiratory failure). The most common adverse reactions were fever/chills (17 patients), urticaria (18 patients), nausea and vomiting, (21 patients), and fatigue (7 patients). Three patients discontinued augmentation therapy because of repeated episodes of fever and chills following infusions from different batches. No deaths or instances of viral transmission (HIV or hepatitis) were observed.
Summary of adverse experiences associated with intravenous augmentation therapy
In a second large series Stoller et al16 reported the frequency and types of adverse experiences among 747 augmentation therapy recipients (Prolastin) in the NHLBI Registry. Using a “severity based” classification system, 174 subjects reported 720 adverse events, the commonest of which were dyspnoea (47%) and dizziness/fainting (17%). Of the adverse events, 8.8% (63/720) were deemed severe, 72.4% (521/720) moderate, and 18.9% (136/720) were deemed mild. Classifying these same adverse events using a “consequence based” classification system showed that only 24% of the 720 adverse events elicited a response—that is, hospital admission or emergency room visit (12/720, 1.7%), physician visit or prescription of a new medication (152/720, 21.1%), or discontinuation of augmentation therapy (8/720, 1.1%).16
Assessment of the frequency of adverse events by infusion frequency (weekly, monthly, other) showed that the absolute frequency of adverse events was very low (95% confidence interval 0.019 to 0.023 events per patient-month), but that the rate was higher among recipients of weekly infusions (0.030 events per patient-month) than among those receiving augmentation therapy with other frequency (0.024 events per patient-month (every 2–3 weeks) or 0.005 events per patient-month (monthly), p<0.001 v weekly rate). However, even at the highest estimated rate of adverse events, participants experienced an adverse event on average only 2.0 times over 5 years of continuous treatment with augmentation therapy. Notably, in these and other studies5 no instance of hepatitis, HIV, or prion disease acquisition has been attributed to intravenous augmentation therapy.
COST EFFECTIVENESS OF INTRAVENOUS AUGMENTATION THERAPY
Intravenous augmentation therapy is expensive (mean annual drug cost estimates of US$28 075–65 973 in 199817,18) and three cost effectiveness studies have been performed to date.19–21 Such analyses must necessarily adopt different assumptions concerning the natural history of the disease, condition of the patients, efficacy of the intervention, cost and duration of augmentation therapy, and other variables. While sensitivity analyses can test the robustness of these assumptions, dissimilarities remain in the fundamental characteristics and conclusions of the studies described here (table 3).
Comparison of cost effectiveness studies of augmentation therapy for AAT deficiency
While the earliest cost effectiveness study by Hay and Robin predates the NHLBI Registry,21 it did analyse cost outcomes under a range of possible therapeutic efficacies and other variables. For instance, for a hypothetical treatment efficacy (that is, reduction in mortality) of 70%, the cost per life-year saved estimates under various assumptions of age, sex, and smoking status ranged from $28 000 to $72 000. For a 40 year old PI*ZZ individual the lowest efficacy at which cost effectiveness would be comparable to that of other common medical interventions was estimated to be 30%, with a cost per life-year saved of $50 ?000–128 000.21 This study estimated a low cost of treatment even after adjustment from 1990 US dollars, did not take into consideration differences in costs and quality of life between health states, and predicted a greater increase in life expectancy and a lower cost per year of life saved to active smokers.
The study by Alkins and O’Malley19 was based on the declining exponential approximation of life expectancy (DEALE), a technique that assumes constant death rates and an inverse relation between remaining life expectancy and annual death rate. Based on the NHLBI Registry data,7 these authors attributed 55% efficacy to augmentation therapy. For individuals with severe AAT deficiency and an FEV1 <50% predicted, the study found that the incremental cost (that is, the value added to usual care) per year of life saved was estimated to be $13 971. The cost calculations spread the cost of augmentation therapy for 5 years over the entire expected increase of life expectancy of 18 years. Also, the study considered only life-years but not quality of life and assessed the costs of augmentation therapy but not those of general COPD treatment such as drugs for COPD other than augmentation therapy.
In the most recent cost effectiveness study, Gildea et al20 conducted analyses that differed from earlier studies in several important respects. Firstly, the authors used a Markov Monte Carlo simulation to assess three different strategies: (1) not treating, (2) treating for life, and (3) treating until the FEV1 is below 35%.20 This simulation used varying transitional probabilities of events over time based on NHLBI Registry data, thereby incorporating known estimates of progression of lung dysfunction and mortality rates into the model. One the main outcome measures was quality adjusted life-years (QALYs), a more comprehensive measure of cost effectiveness than years of life saved. The study also considered the cost of other pulmonary medications used by individuals with AAT deficiency. The incremental cost effectiveness ratio (ICER) was $312 511 per QALY for the lifelong augmentation strategy and $207 841 per QALY for the augmentation therapy until FEV1 <35% strategy. Sensitivity analyses showed that the annual cost of augmentation therapy would have to decrease from $54 765 to $14 000 for the ICER to fall below $100 000. These results differ importantly from those of earlier studies in that the cost of augmentation therapy under a range of plausible conditions remains high and exceeds that of commonly used health interventions.
While there are several significant methodological differences between these studies, one basic difference is whether a constant annual mortality rate is assumed, as with the DEALE method of analysis used in the study by Alkins et al.19 In validation experiments the DEALE method has been found to underestimate the life expectancy compared with that of a more accurate Markov process, particularly at the lower ranges of the excess mortality rates ascribed to AAT deficiency and at the relatively young age of such patients.22
Overall, while the available studies regarding cost effectiveness of augmentation therapy offer widely varying estimates and the most recent results20 suggest less favourable cost effectiveness than other conventional healthcare interventions, it should be emphasised that cost effectiveness considerations should not dominate treatment of approval decisions, especially given the apparent clinical efficacy and the current lack of alternative specific treatments for AAT deficiency. Indeed, any doubts raised about the cost effectiveness of augmentation therapy should encourage the development of alternative even more clinically effective and more cost effective options, as will be discussed in a later article in this series.