Article Text

Abstract
Background: Angiogenesis has been implicated in the pathogenesis of idiopathic interstitial pneumonia (IIP). The aim of this study was to examine the relationship between plasma concentrations of the angiogenic cytokines interleukin 8 (IL-8), vascular endothelial growth factor (VEGF), and endothelin-1 (ET-1) and clinical parameters of disease progression over a 6 month period to identify potential aetiological mediators and prognostic markers of disease activity in patients with IIP.
Methods: Forty nine patients with IIP (40 men) were recruited to the study. Plasma cytokine measurements, pulmonary function tests, and high resolution computed tomography (HRCT) scans were performed on recruitment and after 6 months. Plasma cytokine measurements were also performed in 15 healthy volunteers for control purposes.
Results: Patients with IIP had significantly higher median (IQR) baseline concentrations of IL-8 and ET-1 than controls (155 (77–303) pg/ml v 31 (0–100) pg/ml, p<0.001) and (1.21 (0.91–1.88) pg/ml v 0.84 (0.67–1.13) pg/ml, p<0.01), respectively. Baseline concentrations of IL-8, ET-1, and VEGF were significantly related to the baseline HRCT fibrosis score (r = 0.42, p<0.005; r = 0.39, p<0.01; and r = 0.42, p<0.005, respectively). Patients with IIP who developed progressive disease had significantly higher baseline levels of IL-8 (345 (270–497) pg/ml v 121 (73–266) pg/ml, p = 0.001) and VEGF (1048 (666–2149) pg/ml v 658 (438–837) pg/ml, p = 0.019). Over 6 months the change in VEGF was significantly related to the change in HRCT fibrosis score (r = 0.565, p = 0.035) and negatively related to the change in forced vital capacity (r = −0.353, p = 0.035).
- DIP, desquamative interstitial pneumonia
- ET-1, endothelin-1
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- IIP, idiopathic interstitial pneumonia
- IL-8, interleukin 8
- IPF, idiopathic pulmonary fibrosis
- NSIP, non-specific interstitial pneumonia
- TLC, total lung capacity
- Tlco, carbon monoxide transfer factor
- VEGF, vascular endothelial growth factor
- idiopathic interstitial pneumonia
- pulmonary fibrosis
- angiogenesis
- cytokines
- prognostic markers
Statistics from Altmetric.com
- DIP, desquamative interstitial pneumonia
- ET-1, endothelin-1
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- IIP, idiopathic interstitial pneumonia
- IL-8, interleukin 8
- IPF, idiopathic pulmonary fibrosis
- NSIP, non-specific interstitial pneumonia
- TLC, total lung capacity
- Tlco, carbon monoxide transfer factor
- VEGF, vascular endothelial growth factor
The idiopathic interstitial pneumonias (IIP) are a heterogeneous group of diffuse parenchymal lung diseases. Idiopathic pulmonary fibrosis (IPF) is the most common and lethal form with a median survival of 2.9 years from diagnosis.1 The interstitial pneumonias are characterised by inflammation, cell proliferation, and excessive extracellular matrix deposition,2 and several cytokines—including those involved with angiogenesis—have been implicated in their pathogenesis. The existence of neovascularisation in patients with IPF was originally identified by Turner-Warwick3 who demonstrated the presence of anastomoses between the systemic and pulmonary microvasculature in lung tissue. Peao et al4 later showed major vascular modifications in an animal model of lung fibrosis including neovascularisation in peribronchial regions and distortion of the architecture of alveolar capillaries. These abnormalities were associated with areas of pulmonary fibrosis.
Vessel formation requires endothelial cell migration, proliferation and degradation of extracellular matrix, and its initiation is dependent upon the balance between angiogenic and angiostatic factors. Studies have shown that angiogenic members of the CXC chemokine family—such as interleukin 8 (IL-8) and epithelial neutrophil activing protein 78—have increased expression in fibrotic lung tissue compared with the angiostatic chemokines.5,6 Furthermore, neutralisation of these chemokines reduces bleomycin induced pulmonary fibrosis and is paralleled by a reduction in angiogenesis.7 Vascular endothelial growth factor (VEGF) is a fundamental regulator of normal and pathological angiogenesis.8 It is a specific mitogen for vascular endothelial cells and stimulates the degradation of extracellular matrix.8 VEGF is reported to be abundant in the lung, with alveolar cells being the site of highest immunohistochemical activity and mRNA expression.9 It has been implicated in the pathogenesis of bleomycin induced fibrosis10,11 and altered levels of VEGF have been found in plasma and bronchoalveolar lavage fluid in patients with pulmonary fibrosis compared with normal controls.12–,14 Endothelin-1 (ET-1) belongs to a family of potent vasoactive peptides and has numerous and diverse actions including vasoconstriction, bronchoconstriction, and growth promotion.15 In patients with IPF ET-1 is thought to promote fibroblast and alveolar epithelial cell proliferation, fibroblast differentiation into myofibroblasts, chemotaxis, contraction and collagen synthesis, while inhibiting collagen degradation.16 It is able to induce a number of fibrogenic growth factors through paracrine stimulation of different cell types including tumour necrosis factor-α, transforming growth factor-β, and fibronectin.16 It may also enhance neovascularisation through induction of VEGF.16 Human studies have shown that ET-1 expression is increased in airway epithelium and type II pneumocytes of patients with IPF compared with control subjects,17 and Hocher and colleagues have shown that chronic overexpression of ET-1 causes lung fibrosis in ET-1 transgenic mice.18
Collectively, these studies suggest that angiogenesis has a role in the pathogenesis of pulmonary fibrosis. The aim of this study was therefore to test the hypothesis that expression of angiogenic cytokines is linked to progression of clinical pulmonary fibrosis. This was investigated by examining the relationship between plasma concentrations of the angiogenic cytokines IL-8, VEGF and ET-1 and clinical parameters of disease progression over a 6 month period to identify potential aetiological mediators and prognostic markers of disease activity in patients with IIP.
METHODS
Study population and group classification
Forty nine patients were recruited to this prospective study from the Manchester Interstitial Lung Disease Clinic (a tertiary referral centre receiving referrals from the North West of England) over a 15 month period (March 1998 to June 1999). Of the 150 patients with IIP attending the clinic, 49 were recruited for baseline measurements and 40 patients re-attended for follow up measurements (cytokine measurements n = 40, lung function studies n = 36, HRCT n = 14) 6 months later. Of the nine patients not followed up, two died and seven dropped out of the study.
A diagnosis of IIP was made on the basis of clinical, HRCT, and pulmonary physiological features as described by the Consensus Classification of IIP.19 A lung biopsy was performed from a single lobe when radiological features were not characteristic of a specific interstitial pneumonia. Patients were excluded if any of the following were present: (1) collagen vascular disease, (2) occupational or environmental exposure to organic dusts, (3) ingestion of drug or agent known to cause pulmonary fibrosis, (4) presence of left ventricular failure. The diagnoses included IPF (n = 39), non-specific interstitial pneumonia (NSIP, n = 6), and desquamative interstitial pneumonia (DIP, n = 4). After the 6 month follow up period patients were classified as progressors or non-progressors according to the percentage change in forced vital capacity (⩾10% or <10% reduction, respectively) or by the clinical criteria described in the ATS consensus statement on IPF if lung function data were not available.20 Fifteen healthy individuals who worked in the North West Lung Centre consented to take part as controls. Informed consent was obtained from each participant and the South Manchester ethics committee approved the study protocol.
Pulmonary function
Pulmonary function was assessed on recruitment and 6 months later by measuring the forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC) (Vmax 22; Sensorimedics Yorba Linda, CA, USA), total lung capacity (TLC) (6200 Plethysmograph; Sensorimedics), and carbon monoxide transfer factor (Tlco) (P K Morgan, Rainham, UK). Values were expressed as percentage of predicted values calculated according to sex, height, and age using the European Community for Steel and Coal Classification.21
High resolution CT scanning
HRCT scans were performed at recruitment and after 6 months according to a standard protocol (1.5 mm sections at 10 mm intervals from the lung apices to the bases with a high spatial frequency algorithm; Picker PQ CT, Picker International, Stevenage, UK). Each lobe was scored independently by two specialist thoracic radiologists (AWH and MSG) who were unaware of any clinical or physiological details of the patients, quantifying the amount of fibrosis on a scale of 0–5 as described by Kazerooni et al.22 A single fibrosis score was then calculated by averaging the scores for each lobe.
Preparation of plasma
Ten ml of venous blood were drawn into an ethylene diamine tetra acetic acid (EDTA) bottle using a tourniquet, centrifuged at 300g for 12 minutes, and then the plasma was separated and stored at −70°C.
Cytokine assays
Plasma samples were analysed for IL-8, VEGF, and ET-1 using specific enzyme linked immunosorbent assays (ELISA). The ELISA capture and detection antibodies for assaying IL-8 and VEGF were selected paired reagents optimised for ELISA performance from R&D Systems (Abingdon, UK). The ET-1 assay (QuantiGlo) was obtained from R&D Systems. The detection limits for the assays were as follows: IL-8 10 000–19 pg/ml; VEGF 10 000–19 pg/ml; ET-1 1000–0.32 pg/ml. The plasma samples were undiluted for the IL-8 and VEGF assays and were diluted 1:1 with assay diluent for the ET-1 assay.
Statistical analysis
Data were expressed as median (25–75% interquartile range (IQR)). Analysis was undertaken using SPSS version 10.1 (SPSS Inc, Chicago, IL, USA). Comparisons between two groups were performed using a Mann-Whitney U test. Comparisons between more than two groups were performed using a Kruskal-Wallis test and those comparisons that were statistically significant were further examined using a Mann-Whitney U test. Correlations between different parameters were determined by Spearman’s rank correlation coefficient. p values of <0.05 were regarded as significant. Receiver operating characteristic (ROC) analysis was performed by standard methods.23
RESULTS
The demographic data of the study patients are shown in table 1⇓; 39 patients had IPF, six had NSIP, and four had DIP. The non-IPF patients were younger, had a lower male: female ratio, and had a shorter duration of symptoms. Twenty three patients were recently diagnosed (within 3 months), 10 of whom (43%) had already started treatment (four prednisolone alone and six prednisolone + other immunosuppressants). Of the remaining 26 patients with a longer standing diagnosis of IIP, half were receiving immunosuppressive treatment (six prednisolone alone, four prednisolone + other, and three other combinations of treatment). Other immunosuppressant therapy included azathioprine, cyclophosphamide, cyclosporin, mycophenolate mofetil, and tacrolimus. All of the patients with DIP had been current smokers at the time of initial presentation.
Baseline demographic data, clinical characteristics, and cytokine concentrations
At baseline all patients were grouped together and compared with healthy control subjects. Median (IQR) baseline plasma IL-8 concentrations in patients with IIP were significantly higher than in the controls (155 (77–303) pg/ml v 31 (0–100) pg/ml, p<0.001). Baseline plasma ET-1 levels were also significantly higher in patients with IIP than in controls (1.21 (0.91–1.88) pg/ml v 0.84 (0.67–1.13) pg/ml, p = 0.003; fig 1⇓). There was no significant difference in baseline VEGF concentrations between patients with IIP and controls (672 (452–915) pg/ml v 648 (433–734) pg/ml, p = 0.257).

Baseline plasma cytokine concentrations in patients with idiopathic interstitial pneumonia (IIP) and controls.
There were no significant differences in baseline plasma cytokine concentrations between the three histological subgroups (data not shown), nor were there any significant differences between baseline plasma cytokine concentrations in the IPF group and in the NSIP and DIP groups combined (data not shown). There were no significant differences in plasma cytokine concentrations at baseline and at 6 months between patients receiving or not receiving treatment (data not shown).
Plasma cytokine concentrations were correlated with lung function parameters and the HRCT fibrosis score at baseline. There was a significant positive relationship between the HRCT fibrosis score and plasma concentrations of IL-8 (r = 0.42, p = 0.0035), ET-1 (r = 0.39, p = 0.007), and VEGF (r = 0.42, p = 0.0035; fig 2⇓). There were also significant negative relationships between the percentage predicted FVC and the plasma concentration of ET-1 (r = −0.30, p<0.05), and the percentage predicted Tlco and the plasma concentration of IL-8 (r = −0.37, p<0.05).

Baseline relationship between HRCT fibrosis score and plasma concentrations of (A) ET-1, (B) IL-8, and (C) VEGF.
In order to assess whether plasma cytokine concentrations at baseline reflected future disease activity, after the 6 month follow up period patients were divided into progressor and non-progressor groups, as described earlier. Of the 49 patients recruited at baseline, 13 did not have lung function measurements repeated at 6 months. Three patients were too breathless to have lung function studies performed (but had blood taken for cytokine measurements), two patients died of progressive disease, one refused lung function tests, and seven dropped out of the study. These 13 patients were classified as progressors (n = 5) and non-progressors (n = 8) according to their clinical course as described by the ATS consensus statement.20 There were 10 progressors and 39 non-progressors. Nine of the progressors had IPF and one had NSIP. Median (25–75% IQR) concentrations of plasma IL-8, ET-1 and VEGF at baseline and after 6 months in progressor and non-progressor groups are shown in table 2⇓. Plasma levels of IL-8, ET-1, and VEGF were higher in progressors than in non-progressors at baseline and 6 months. This was statistically significant for IL-8 at both time points, and for VEGF at baseline and ET-1 at 6 months.
Plasma cytokine concentrations (pg/ml) in progressors (P) and non-progressors (NP) at baseline and at 6 months
ROC analysis was used to evaluate the diagnostic value of baseline plasma IL-8, VEGF and ET-1 concentrations in predicting future disease progression (fig 3⇓). The area under the curve was 0.833 (95% CI 0.69 to 0.98) for IL-8, 0.741 (95% CI 0.54 to 0.94) for VEGF, and 0.692 (95% CI 0.67 to 0.95) for ET-1.

Receiver operator characteristic curves for identification of patients with progressive disease.
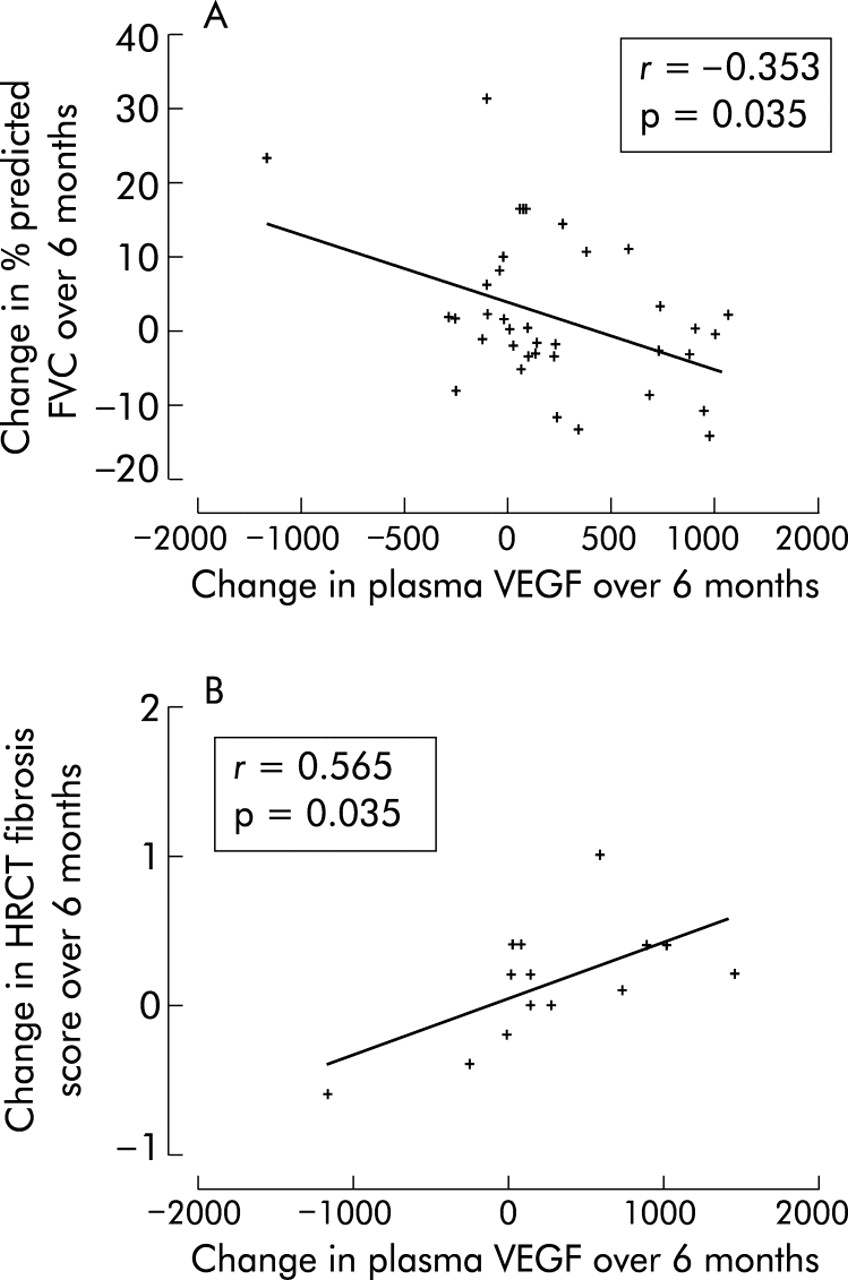
The relationship between the change in plasma cytokine concentrations and the change in clinical variables over 6 months was also examined. The statistically significant findings were a positive relationship between the change in HRCT fibrosis score and the change in plasma VEGF (r = 0.565 p = 0.035; fig 4⇓) and a negative relationship between the change in percentage predicted FVC and the change in plasma VEGF (r = −0.353 p = 0.035; fig 4⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlates of change in plasma VEGF over 6 months.
DISCUSSION
This study on plasma cytokine levels was carried out in a large heterogeneous group of patients with IIP attending a tertiary referral service. We have shown that patients with IIP have significantly higher baseline plasma concentrations of IL-8 and ET-1 but not VEGF than controls. We have shown for the first time that the baseline plasma concentrations of all three cytokines correlate with the baseline HRCT fibrosis score, which is known to relate to prognosis and survival.24 Patients with IIP who developed progressive disease had significantly higher baseline plasma levels of IL-8 and VEGF. Furthermore, a rising VEGF concentration was associated with an adverse physiological outcome, as suggested by lung function and HRCT parameters.
These data from a heterogeneous group of patients are not confounded by type of IIP or by treatment since there were no differences between subgroups. However, more data are needed in the smaller subgroups to confirm these observations. These data also suggest that the fibrotic component of NSIP and IPF is unresponsive to current treatments and might have a final common pathway leading to fibrosis. Histological studies support a final common pathway as areas of both NSIP and DIP have been found on a background of typical IPF.25,26
The raised plasma levels of these angiogenic cytokines suggest that they have a key role in the progression of IPF and that a proangiogenic environment co-exists with progressive fibrosis. Specifically, an important role of ET-1 has been suggested by the attenuation of bleomycin induced pulmonary fibrosis in rats by the ET-1 receptor antagonist Bosentan.27 Our study supports the concept of ET-1 as a potential therapeutic target. Secondly, the sequential VEGF data support the concept that VEGF receptor II antagonists, which are in phase II trials as tumour angiogenesis inhibitors, may be a useful additional therapy for patients with progressive pulmonary fibrosis.
Recent clinical guidelines for the management of IPF recommend the use of potent immunosuppressive agents (such as prednisolone, azathioprine, and cyclophosphamide) with generally poor response rates and substantial side effect profiles.20 Many patients with IPF are elderly and have stable or only slowly progressive disease. In these circumstances, commencing treatment is a difficult decision28 and it would be helpful to have simple biological prognostic markers to predict those patients who will deteriorate. The identification of progressive disease by baseline plasma IL-8 and VEGF levels and also by sequential plasma VEGF levels is a potentially important clinical advance. For example, the ROC curve analysis suggested that the baseline plasma IL-8 concentration was sensitive and specific at identifying patients with progressive disease; a cut off value of 296 pg/ml had a sensitivity of 80% and specificity of 85%. This needs reproducing in further studies, but it may be possible to devise a biological prediction index based on a combination of different plasma markers. These could enable us to target treatment to those patients who need it and avoid toxicity in those patients who do not.
This study has a number of limitations. Firstly, the study population was heterogeneous and included those with IPF, NSIP and DIP. Furthermore, participants were at different stages of their disease and were receiving different treatments. These methodological compromises were necessary to generate a study cohort of sufficient size. Furthermore, it enabled us to determine the cytokine profiles of three different IIP subgroups. The results were not significantly altered by considering the IPF group in isolation and there were no differences in the plasma cytokine concentrations between patients prescribed and not prescribed treatment. Secondly, the patients and controls were not well matched for age and this reflects our difficulty in recruiting non-diseased elderly controls. It is not known whether age affects plasma concentrations of these angiogenic cytokines and it is possible that this age difference explains why plasma VEGF levels were not significantly different between patients and controls, yet correlated with the HRCT fibrosis score. This illustrates the need for control data from elderly non-diseased individuals. In our opinion, the relationship between the baseline VEGF concentration and the HRCT fibrosis score is robust and is supported by the fact that patients who later developed progressive disease had higher baseline VEGF concentrations than those who remained stable. Thirdly, biopsy samples were not taken from three of the non-IPF patients. Although the clinical and radiological features were considered diagnostic at the time, a recent study has shown that a diagnosis of NSIP based on HRCT was correct in only 41% of cases.29 Fourthly, the IL-8 and ET-1 concentrations were higher in the progressors than in the non-progressors at baseline and at 6 months, but their concentrations declined with time. This was an unexpected finding and might be explained by the reduced number of patients studied at follow up compared with baseline, or might reflect the effect of end stage fibrotic disease. Fifthly, only 14 of 40 patients had a repeat HRCT scan at 6 months so the longitudinal HRCT data must be interpreted with care. Finally, although the findings of this paper suggest that angiogenic cytokines have a role in the progression of IIP, it is also possible that these relationships are epiphenomena and simply reflect new vessel formation associated with a bad fibrotic process that is driven by other factors. Despite these limitations, we feel that our findings provide novel data about potential mediators of lung fibrosis in the human model that are of aetiological and prognostic significance.
In conclusion, this is the first longitudinal study evaluating serial angiogenic cytokine measurements in patients with IIP and the results strengthen the assertion that angiogenic cytokines are involved in the fibrotic pathway. Our data also suggest that non-invasively acquired markers deserve further evaluation in terms of their potential to identify patients who are likely to develop progressive disease, so that treatment can be more effectively targeted. Such markers might also be used to monitor the response to treatment.
REFERENCES
Supplementary materials
Publisher Correction
Please note that the Conclusions are missing from the abstract of this article.
The missing text is shown below:
Conclusions: These results support a role for angiogenesis in the pathogenesis of IIP, and IL-8 and VEGF deserve further evaluation as prognostic markers.The complete abstract is provided as a PDF here.
The error is much regretted.
Files in this Data Supplement: