Article Text

Abstract
Background: Individuals with chronic obstructive pulmonary disease (COPD) are at increased risk of cardiovascular diseases, osteoporosis, and muscle wasting. Systemic inflammation may be involved in the pathogenesis of these disorders. A study was undertaken to determine whether systemic inflammation is present in stable COPD.
Methods: A systematic review was conducted of studies which reported on the relationship between COPD, forced expiratory volume in 1 second (FEV1) or forced vital capacity (FVC), and levels of various systemic inflammatory markers: C-reactive protein (CRP), fibrinogen, leucocytes, tumour necrosis factor-α (TNF-α), and interleukins 6 and 8. Where possible the results were pooled together to produce a summary estimate using a random or fixed effects model.
Results: Fourteen original studies were identified. Overall, the standardised mean difference in the CRP level between COPD and control subjects was 0.53 units (95% confidence interval (CI) 0.34 to 0.72). The standardised mean difference in the fibrinogen level was 0.47 units (95% CI 0.29 to 0.65). Circulating leucocytes were also higher in COPD than in control subjects (standardised mean difference 0.44 units (95% CI 0.20 to 0.67)), as were serum TNF-α levels (standardised mean difference 0.59 units (95% CI 0.29 to 0.89)).
Conclusions: Reduced lung function is associated with increased levels of systemic inflammatory markers which may have important pathophysiological and therapeutic implications for subjects with stable COPD.
- COPD, chronic obstructive pulmonary disease
- CRP, C-reactive protein
- FEV1, forced expiratory volume in 1 second
- IL, interleukin
- TNF-α, tumour necrosis factor-α
- chronic obstructive pulmonary disease
- inflammation
- C-reactive protein
- meta-analysis
Statistics from Altmetric.com
- COPD, chronic obstructive pulmonary disease
- CRP, C-reactive protein
- FEV1, forced expiratory volume in 1 second
- IL, interleukin
- TNF-α, tumour necrosis factor-α
Systemic inflammation is increasingly being recognised as a risk factor for a number of different complications including atherosclerosis,1 cachexia,2 anorexia,2,3 and osteoporosis.4 Notably, all of these complications are commonly observed in patients with chronic obstructive pulmonary disease (COPD).5–,10 Whether systemic inflammation is present in stable COPD and whether it is wholly or partially responsible for these associations is controversial. Although several studies have been undertaken to evaluate this potential relationship, most of the studies have been small in size and scope and, as such, may on their own have lacked sufficient statistical power to address this issue adequately. To overcome these and other limitations and to understand better the relationship between COPD and systemic inflammation, we conducted a systematic review and meta-analysis with the specific aim of examining the associations between stable COPD and serum levels of C-reactive protein (CRP), fibrinogen, leucocytes, and various proinflammatory cytokines. We chose these markers of systemic inflammation because they have been well studied and have been intimately linked with the development of ischaemic heart disease and stroke11–,13 which, interestingly, are also the leading causes of mortality among patients with COPD.14
METHODS
Search for relevant studies
Using MEDLINE (1966–2003), EMBASE, CINAHL (1982–2003), and the Cochrane databases we conducted a systematic literature search to identify relevant studies published before 1 November 2003 which evaluated the potential relationship between stable COPD and various markers of systemic inflammation. Disease-specific search terms (COPD, bronchitis, emphysema, forced expiratory volume, or vital capacity) were combined with inflammatory marker-specific search terms (systemic inflammation, biological markers, C-reactive protein, fibrinogen, leucocyte, interleukin, interleukin-8, interleukin-6, or tumour necrosis factor-α) in all our searches. The electronic searches were supplemented by scanning the reference lists from retrieved articles to identify additional studies that may have been missed during the initial search. We also contacted the primary authors for additional data and/or clarification of data, when necessary, to ensure that all relevant articles were represented in the meta-analysis. It was decided a priori to include only those studies in which stable patients (or individuals) were studied. All acute exacerbation studies were therefore discarded, as were those that did not have a suitable comparator group.
Study selection and data abstraction
The primary outcome of this systematic review was to compare serum CRP, fibrinogen, leucocyte, tumour necrosis factor (TNF)-α, interleukin 6 (IL-6), and interleukin 8 (IL-8) levels between study participants with and without stable COPD. From each relevant article two investigators (WQG, DDS) abstracted the following information: source of the data, study design, baseline characteristics of study participants including age, predicted forced expiratory volume in 1 second (FEV1), and smoking status. We also evaluated the laboratory methods used to determine the levels of systemic inflammatory markers. Any questions or discrepancies regarding these data were resolved through iteration and consensus. We used the definition of COPD provided by each individual study, however they were defined. Although there was some heterogeneity in the way in which COPD was defined across the studies, most defined COPD using spirometric criteria of FEV1/forced vital capacity (FVC) <0.70 or <0.60. For population based studies in which COPD was not explicitly defined, we assumed that participants in the lowest quartile group of predicted FEV1 had COPD while those in the highest quartile group of predicted FEV1 did not (and therefore served as controls). We assumed that most individuals in the former category had COPD since, from a population perspective, COPD is the most common cause of chronic airflow limitation in the adult population. For these studies we included data only from those participants who had a history of smoking; data from lifetime non-smokers were censored from the main analyses. Studies which did not provide spirometric data on the study participants were excluded to ensure comparability of the COPD definition across the studies.
Statistical methods
To accommodate differences in the way in which inflammatory markers were measured and reported across various laboratories, the absolute levels of the above inflammatory markers were converted into a common unit by calculating standardised effect sizes. Standardised effect sizes were derived by dividing the mean difference in CRP levels between COPD and control subjects of each study by its standard deviation.15 The same technique was used to calculate standardised mean differences for leucocytes, fibrinogen, and other inflammatory cytokines. For each outcome we tested the heterogeneity of results across the studies using a Cochran Q test. If significant heterogeneity was observed (p<0.10), a random effects model—which assigns a weight to each study based on individual study variance as well as between study variance—was used to pool the results together. In the absence of significant heterogeneity a fixed effects model was used.16 As a sensitivity analysis we also pooled the data together using a weighted mean difference technique. All analyses were conducted using Review Manager version 4.2 (Revman; The Cochrane Collaboration, Oxford, UK).
RESULTS
A summary of the search strategy is shown in fig 1⇓. The original search yielded 911, 666, 279, and 16 citations in MEDLINE, EMBASE, CINAHL, and the Cochrane Databases, respectively. The abstracts of these articles were selected and reviewed. Of these, 19 articles were retrieved for a detailed review: seven for CRP,17–,23 six for fibrinogen,17,20,24–,27 six for leucocytes,17,18,20,28–,30 six for TNF-α,22,31–,35 two for IL-6,22,23 and two for IL-8.23,35 Five studies were excluded for the following reasons: two studies were publications of the same cohort;17,34 two provided data on leucocytes based only on a linear regression model which made it impossible to ascertain the relationship between COPD and leucocytes;28,30 and one study diagnosed chronic bronchitis based only on symptoms (without spirometry).27 This process left 14 original studies meeting the inclusion and exclusion criteria which were then used for the analyses: five for CRP,18–,22 four for fibrinogen,20,24–,26 four for TNF-α,22,31–,33 three for leucocytes,18,20,29 two for IL-8,23,35 and one for IL-6.22 The relevant baseline data from each of the selected studies are summarised in table 1⇓.
Baseline information on individual studies included in the meta-analysis

Study selection process.
Patients with COPD had higher levels of CRP than control subjects in all studies. Overall, the standardised mean difference in the CRP level between COPD and control subjects was 0.53 units (95% CI 0.34 to 0.72; fig 2⇓) or 1.86 mg/l (95% CI 0.75 to 2.97) using a weighted mean difference technique. The heterogeneity in results across the studies (test for heterogeneity, p = 0.006) probably resulted from differences in severity of the underlying COPD population and ways in which control subjects were selected for each study. Mannino et al20 and Mendall et al,21 for instance, used data from a population based study whereas Eid et al19 and Yasuda et al22 recruited their patients from respiratory clinics. Not surprisingly, the standardised mean difference values in CRP were larger in the latter studies than the former. Importantly, however, even in population based studies, which are less susceptible to selection bias, a strong relationship was observed between CRP and COPD which suggests that COPD is, indeed, a risk factor for increased CRP in the community.

Relationship between C-reactive protein (CRP) and COPD. Continuous variables are expressed as mean (SD) unless otherwise specified. *Imputed from the regression coefficient between mean FEV1 (25–75th percentile) and CRP.
Similarly, patients with COPD had higher fibrinogen levels than control subjects. Overall, the standardised mean difference in the fibrinogen level was 0.47 units (95% CI 0.29 to 0.65, fig 3⇓) or 0.37 g/l (95% CI 0.18 to 0.56) using a weighted mean difference technique. As with the CRP results, there was some heterogeneity in the results between the studies (test for heterogeneity, p<0.0001). However, all studies (both large and small) showed that fibrinogen levels were higher in COPD than in control subjects. For population based studies20,25,26 the standardised mean difference between the lowest quartile group and the highest quartile group of predicted FEV1 among smokers was 0.43 units (95% CI 0.24 to 0.61).25,26
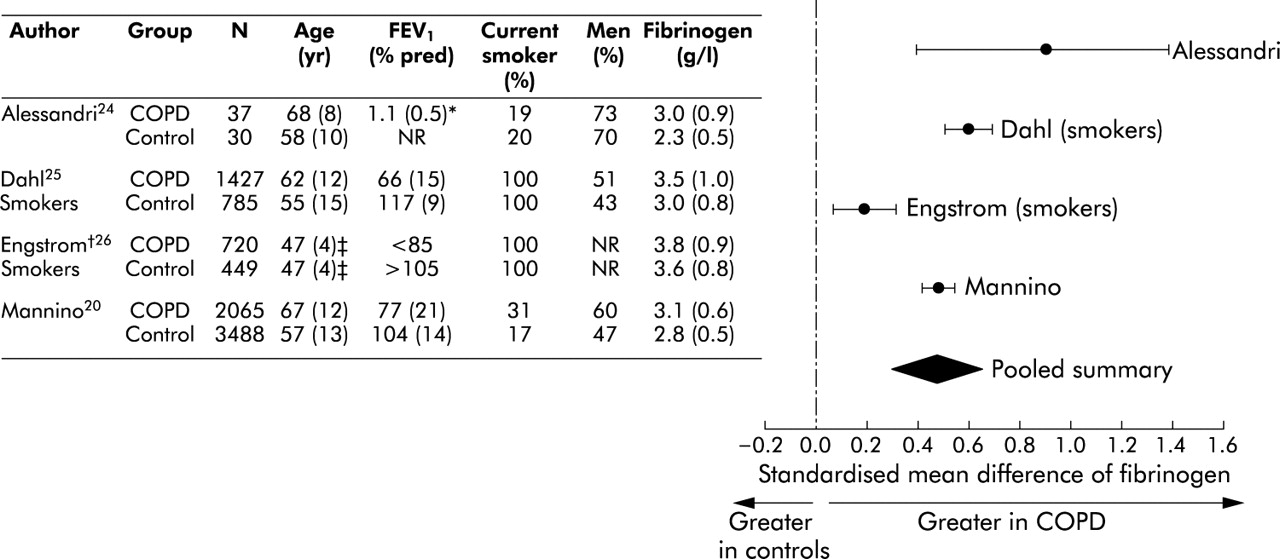
Relationship between fibrinogen and COPD. Continuous variables are expressed as mean (SD) unless otherwise specified. *FEV1 in litres. †Based on forced vital capacity. ‡Estimated.
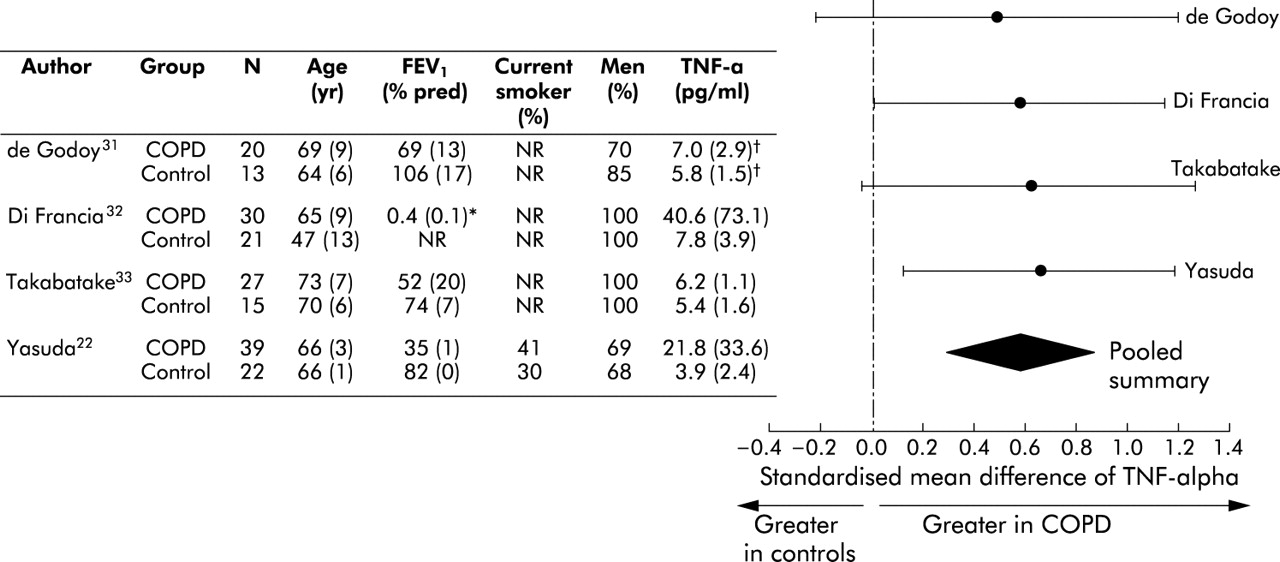
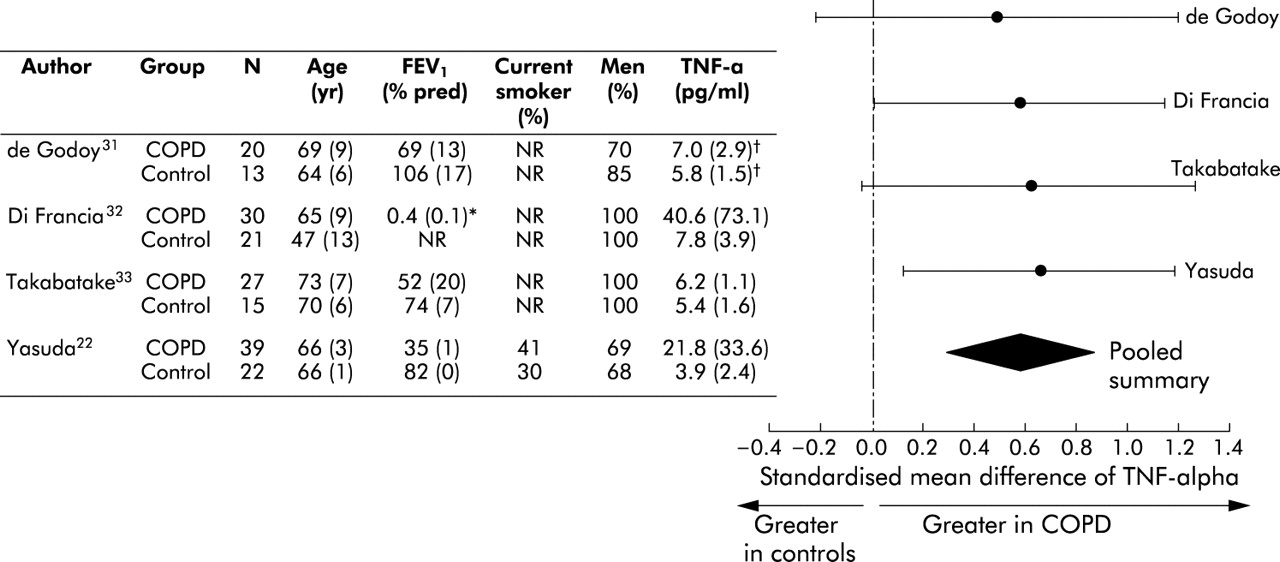
Overall, circulating leucocytes were higher in patients with COPD than in control subjects. The standardised mean difference was 0.44 units (95% CI 0.20 to 0.67; test for heterogeneity, p = 0.003) or 0.88×109 cells/l (95% CI 0.36 to 1.40) using a weighted mean difference technique (fig 4⇓). Likewise, serum TNF-α levels were higher in patients with COPD than in control subjects. The standardised mean difference was 0.59 units (95% CI 0.29 to 0.89; test for heterogeneity, p = 0.87) or 2.64 pg/ml (95% CI −0.44 to 5.72) using a weighted mean difference technique (fig 5⇓).

Relationship between leucocytes and COPD. Continuous variables are expressed as mean (SD) unless otherwise specified. *Data from smokers only. WBC = white blood cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relationship of tumour necrosis factor-α and COPD. *FEV1/FVC. †Deduced from medians and ranges.
There was only one study with analysable data for IL-6.22 Compared with healthy controls (N = 22), patients with COPD (N = 39) had significantly raised serum levels of IL-6 (mean difference 13.10 pg/ml; 95% CI 7.23 to 18.97).
There were two studies on IL-8.23,35 One study showed that 17 of 30 patients with COPD had a detectable IL-8 level whereas none of the 26 healthy controls had detectable serum IL-8 (using an assay with a detectable limit of 20 pg/ml).23 Another study reported that four out of 18 patients with COPD had detectable IL-8 levels in their serum compared with none of 17 healthy controls (using an assay with detectable limit of 8 pg/ml).35
DISCUSSION
In this systematic review we found that, compared with healthy controls, individuals with chronic airflow limitation had significantly raised levels of CRP, fibrinogen, leucocytes, and TNF-α, indicating that persistent systemic inflammation is present in COPD. Even among non-current smokers there was evidence for low grade systemic inflammation in those with chronic airflow limitation. This suggests that, once COPD develops, cessation of smoking may not fully attenuate the inflammatory process associated with this condition.
How and why individuals with COPD develop systemic inflammation is uncertain and unknown. COPD is characterised by an intense inflammatory process in the airways, parenchyma, and pulmonary vasculature.36 It is possible in some cases that the inflammatory process may “spill” over into the systemic circulation, promoting a generalised inflammatory reaction.37–,40 It is also possible that there are common genetic or constitutional factors that may predispose individuals with COPD to both systemic and pulmonary inflammation.36,41 Furthermore, while we believe that COPD is responsible for the systemic inflammation, there exists the possibility of reverse causation. The possibility that systemic inflammation causes injuries to the airways leading to COPD changes cannot be fully discounted.25
Whatever the mechanism, the presence of systemic inflammation in COPD has been linked with a variety of complications including weight loss,19,31,32 cachexia,8,10 osteoporosis,9,10 and cardiovascular diseases.5–,7 Moreover, data from Dahl et al suggest that individuals with increased systemic inflammatory markers such as fibrinogen experience an accelerated decline in lung function and are at increased risk of COPD hospitalisations in the future.25 The relationship between COPD, systemic inflammation, and cardiovascular diseases may be especially germane as over half of patients with COPD die from cardiovascular causes.42,43 Indeed, airflow limitation doubles the risk of cardiovascular mortality independent of smoking.5–7,17,26 Moreover, during periods of exacerbation, plasma levels of fibrinogen and serum levels of IL-6 increase significantly, which may further contribute to increased cardiovascular morbidity and mortality in patients with COPD.44
Several limitations of this study should be emphasised. Firstly, all relevant studies regarding the association between impaired lung function and systemic inflammation were cross sectional in nature, so the temporal relationships between these two factors were unclear. Secondly, there was some variation in the way in which inflammatory markers were sampled and analysed. There was also heterogeneity in the sample populations across the studies. Even within the COPD group, some were selected on the basis of weight loss or poor nutritional status and, as such, may not represent the general pool of COPD patients. However, despite these variations, it was reassuring that in nearly all studies (regardless of sample size, baseline FEV1, and composition of the study and control groups), those with airflow limitation had, on average, higher levels of systemic inflammatory markers than healthy controls. This suggests that selection and sampling biases were unlikely to be responsible for the observed associations. Thirdly, there was a marked paucity of studies that evaluated the relationship between COPD and IL-6 or IL-8. IL-6 has been implicated in the pathogenesis of atherosclerosis45–,47 while IL-8 may be an important signalling molecule for neutrophils which may have significance in the pathophysiology of COPD.48,49 In view of their potential relevance in COPD, more studies are needed in the future to determine whether the systemic expression of these cytokines is increased in COPD.
In summary, there is now a large body of evidence to indicate that systemic inflammation is present in patients with stable COPD. This finding may explain, at least in part, the high prevalence of systemic complications such as cachexia, osteoporosis, and cardiovascular diseases among patients with COPD. Future studies are needed to determine whether attenuation of the systemic inflammatory process can modify the risk of these complications in COPD.
Acknowledgments
The authors thank Dr Morten Dahl, Dr Matthew Knuiman and the Busselton Health Study, and Dr Mieke Dentener for providing additional data for this study.
REFERENCES
Footnotes
DDS is supported by a New Investigator Award from the Canadian Institutes of Health Research and by the Glaxo-Smith-Kline/St Paul’s Hospital Foundation COPD Professorship
Linked Articles
- airwaves