Article Text

Abstract
The molecular basis of α1-antitrypsin deficiency is reviewed and is shown to be due to the accumulation of mutant protein as ordered polymers within the endoplasmic reticulum of hepatocytes. The current goals are to determine the cellular response to polymeric α1-antitrypsin and to develop therapeutic strategies to block polymerisation in vivo.
- α1-antitrypsin deficiency
- molecular pathophysiology
Statistics from Altmetric.com
Alpha1-antitrypsin (AAT) deficiency was reported in an Alaskan girl who died 800 years ago1 and may have accounted for the premature death of Frederic Chopin in 1849.2,3 It was first described as a clinical entity in 1963 by Laurell and Eriksson who noted an absence of the α1 band on serum protein electrophoresis.4 The major function of AAT is to protect the tissues against the enzyme neutrophil elastase.5,6 Its role in protecting the lungs against proteolytic attack is underscored by the association of plasma deficiency with early onset panacinar emphysema.7 This finding, together with the observation that the intrapulmonary instillation of elastolytic enzymes also results in emphysema,8–11 gave rise to the proteinase-antiproteinase hypothesis of lung disease. In health there is a balance between proteinases and antiproteinases, but when proteinases are in excess, tissue destruction will ensue. The proteinase-antiproteinase hypothesis was developed over 35 years ago and still remains central to our understanding of the pathogenesis of lung disease. We review here the molecular mechanisms that underlie AAT deficiency and show how an understanding of this mechanism has allowed us to explain the deficiency of other members of the serine proteinase inhibitor or serpin superfamily. These include the deficiency of antithrombin, C1 inhibitor, α1-antichymotrypsin, and neuroserpin in association with thrombosis, angio-oedema, airflow obstruction, and dementia, respectively. We have grouped these conditions together as the “serpinopathies”.12–14 Their common pathophysiology provides a platform for the development of strategies to treat the associated clinical syndromes.
STRUCTURE AND FUNCTION OF α1-ANTITRYPSIN (AAT)
AAT is a 394 amino acid, 52 kDa, acute phase glycoprotein encoded on chromosome 14q31–32.1.15–17 It is synthesised by hepatocytes18,19 and secreted into the plasma at a concentration of 1.9–3.5 mg/ml. It is also synthesised by and secreted from macrophages20 and intestinal21 and bronchial epithelial cells.22 The protein was originally named because of its ability to inhibit pancreatic trypsin.23 Subsequently it has been found to be an effective inhibitor of a variety of other proteinases including neutrophil elastase,5 cathepsin G,5 and proteinase 3.24 The broad spectrum of proteinase inhibition gave rise to its alternative name of α1-proteinase inhibitor,25 although this too is inaccurate as other proteins in the α1 band of serum (such as α1-antichymotrypsin) are also proteinase inhibitors.
Crystal structures have shown that AAT is composed of three β-sheets (A–C) and an exposed mobile reactive loop (fig 1) that presents a peptide sequence as a pseudosubstrate for the target proteinase.26–30 The critical amino acids within this loop are the P1–P1′ residues, methionine serine, as these act as a “bait” for neutrophil elastase.31 After docking, the enzyme cleaves the P1–P1′ peptide bond of AAT32 and the proteinase is inactivated by a mousetrap action (fig 1) that swings it from the upper to the lower pole of the protein in association with the insertion of the reactive loop as an extra strand in β-sheet A.33–37 This altered conformation of AAT bound to its target enzyme is then recognised by hepatic receptors and cleared from the circulation.38

α1-antitrypsin can be considered to act as a mousetrap.26,37,138 Following docking (left), the neutrophil elastase (grey) is inactivated by movement from the upper to the lower pole of the protein (right). This is associated with insertion of the reactive loop (red) as an extra strand into β-sheet A (green). Reproduced from Lomas and Carrell12 with permission.
The remarkable mouse trap action of AAT is central to its role as an effective inhibitor of serine proteinases. Paradoxically, it is also its Achilles heel as point mutations in these mobile domains make the molecule vulnerable to aberrant conformational transitions such as the one that underlies AAT deficiency.
α1-ANTITRYPSIN (AAT) DEFICIENCY
AAT deficiency is the most widely recognised abnormality of a proteinase inhibitor that causes lung disease. Over 70 naturally occurring variants have been described and characterised by their migration on isoelectric focusing gels—the proteinase inhibitor or Pi system.39 The commonest deficiency variants, S and Z, result from point mutations in the AAT gene40–42 and are so named as they make the protein migrate more slowly than normal M AAT. Mutations that cause more rapid migration of AAT are labelled A to L.
A recent review of 70 surveys has provided an assessment of the frequency and distribution of the S and Z AAT alleles throughout Europe.43 The greatest frequency of the S allele occurs within the Iberian peninsula and gradually reduces in the direction of south to north and from west to east. S AAT (264glutamic acid→valine) is found in up to 28% of southern Europeans and, although it results in plasma AAT levels that are 60% of the M allele, it is not associated with any pulmonary sequelae. In contrast, the Z allele is most common in northwest Europe with frequencies declining from west to east and from north to south. The Z variant (342glutamic acid→lysine) results in a more severe deficiency that is characterised in the homozygote by plasma AAT levels of 10% of the normal M allele and 60% in the MZ heterozygote (50% from the M allele and 10% from the Z allele). The Z mutation results in the accumulation of AAT as inclusions in the rough endoplasmic reticulum of the liver.44 These inclusions predispose the homozygote to juvenile hepatitis, cirrhosis,45,46 and hepatocellular carcinoma.47 Furthermore, the lack of circulating protein predisposes the homozygote to early onset panlobular emphysema.7,48,49
MOLECULAR PATHOLOGY OF THE LIVER DISEASE ASSOCIATED WITH PI Z AAT
There is now overwhelming evidence that the liver disease associated with the Z variant of AAT is due to the accumulation of aggregated protein rather than a plasma deficiency. Strong support is provided by the recognition that null alleles, which produce no AAT, are not associated with cirrhosis.39 Moreover, the overexpression of Z AAT in animal models results in liver damage.50,51 Our understanding of the molecular basis of AAT deficiency came from a recognition that the normal active protein undergoes a profound conformational transition to inhibit its target proteinase, neutrophil elastase (see above). The Z mutation of AAT is at residue P17 (17 residues proximal to the P1 reactive centre) at the head of strand 5 of β-sheet A and the base of the mobile reactive loop (fig 2). The mutation opens β-sheet A, thereby favouring the insertion of the reactive loop of a second AAT molecule to form a dimer.26,52–54 This can then extend to form polymers that tangle in the endoplasmic reticulum of the hepatocyte to form inclusion bodies (fig 3). Support for this came from the demonstration that purified plasma Z AAT formed chains of polymers when incubated under physiological conditions.52 The rate of polymer formation was accelerated by raising the temperature to 41°C and could be blocked by peptides that competed for annealing to β-sheet A.52,55 The role of polymerisation in vivo was confirmed by the finding of AAT polymers in inclusion bodies from the liver of a Z AAT homozygote with cirrhosis52,56 and in hepatic cell lines expressing the Z variant.57 Moreover, point mutations that block polymerisation increased the secretion of mutants of AAT from a Xenopus oocyte expression system.58

The structure of α1-antitrypsin (AAT) is centred on β-sheet A (green) and the mobile reactive centre loop (red). Polymer formation results from the Z variant of AAT (Glu342Lys at P17, arrowed) or the Siiyama, Mmalton, S or I mutations in the shutter domain (blue circle) that open β-sheet A to favour partial loop insertion (step 1) and the formation of an unstable intermediate (M*).59,63 The patent β-sheet A can either accept the loop of another molecule (step 2) to form a dimer (D) which then extends into polymers (P)26,52,54 or its own loop (step 3) to form a latent conformation (L).139,140 The individual molecules of AAT within the polymer are coloured red, yellow and blue. Reproduced from Gooptu et al59 with permission.

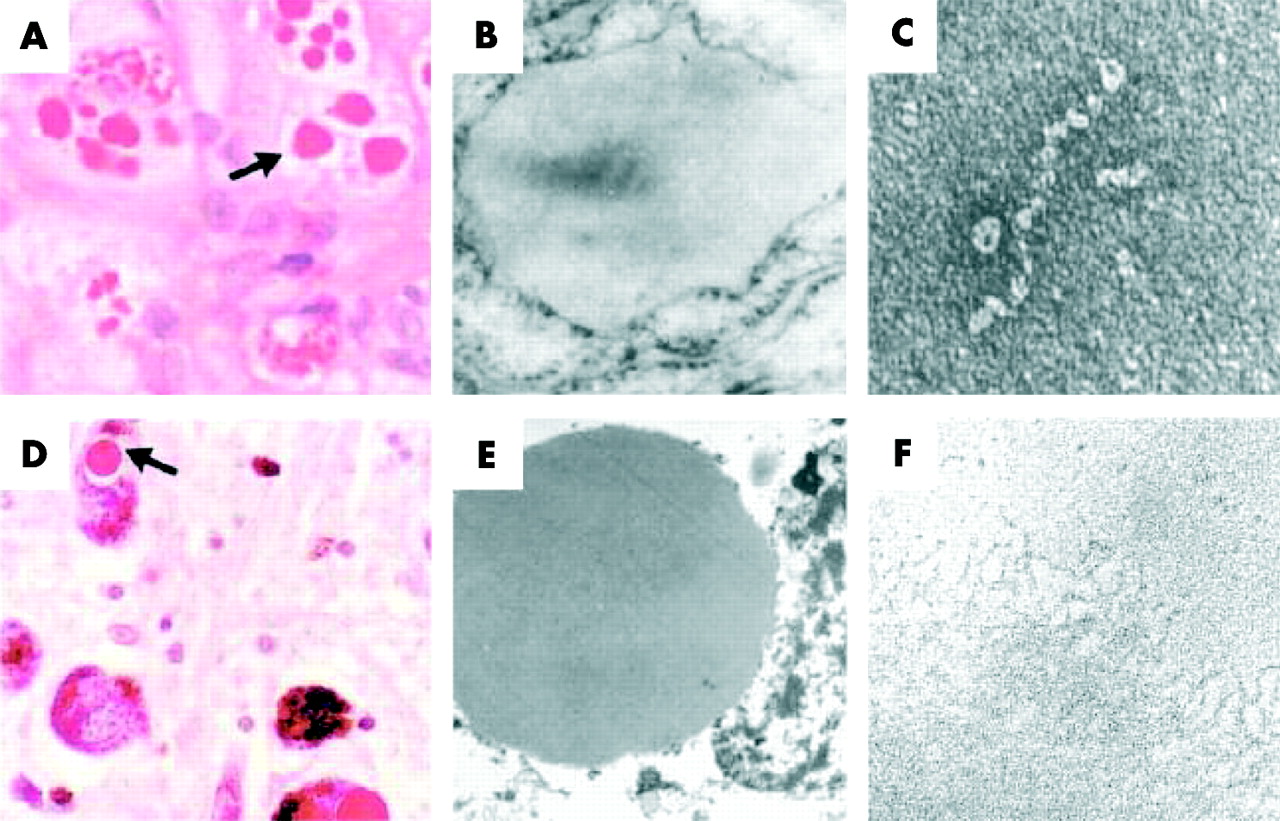
Z α1-antitrypsin (AAT) is retained within hepatocytes as intracellular inclusions. (A) These inclusions are PAS positive and diastase resistant (arrow) and are associated with neonatal hepatitis and hepatocellular carcinoma. (B) Electron micrograph of a hepatocyte from the liver of a patient with Z AAT deficiency shows the accumulation of AAT within the rough endoplasmic reticulum. These inclusions are composed of chains of AAT polymers shown here from the plasma of a Siiyama AAT homozygote (C) and from the liver of a Z AAT homozygote (F). Similar mutations in AAT and neuroserpin result in similar intracellular inclusions of AAT and neuroserpin shown here in (A) hepatocytes and (D) neurones with PAS staining, and as endoplasmic aggregates of the abnormal proteins on electron microscopy (B and E, respectively). Electron microscopy confirms that the abnormal neuroserpin forms bead-like polymers and entangled polymeric aggregates identical to those shown here with Z AAT (C and F, respectively). Magnification left to right: ×200, ×20 000, ×220 000). Reproduced from Carrell and Lomas14 with permission.
The pathway of AAT polymerisation has been assessed by biochemical, biophysical, and crystallographic analysis and is shown in fig 2.53,59 Step 1 represents the conformational change of AAT to a polymerogenic monomeric form (M*), step 2 represents the formation of polymers (P), and step 3 represents a side pathway which leads to the formation of the stable monomeric latent conformation (L). The Z mutation causes most of the unstable protein to form polymers. The presence of the unstable polymerising intermediate M* was predicted from the biophysical analysis of polymer formation,53 the demonstration of an unfolding intermediate,60–62 and solving the crystal structure of a polymerogenic mutant of α1-antichymotrypsin.59 Our latest data suggest that the Z mutation forces AAT into a conformation that approximates the unstable M* and hence favours polymer formation.63
The quality control mechanisms within the hepatocyte that handle polymers are now being elucidated.64–66 Elegant studies have shown that it is the trimming of asparagine linked oligosaccharides that target Z AAT polymers into an efficient non-proteosomal disposal pathway within hepatocytes. However, the proteosome has an important role in metabolising Z AAT in some hepatic67 and extrahepatic68,69 mammalian cell lines. Moreover, there is increasing evidence that the retained Z AAT stimulates an autophagic response within the hepatocyte.70,71 Despite our increased understanding of the disposal pathway, it still remains unclear how the accumulation of Z AAT causes cell death and liver cirrhosis.
The temperature and concentration dependence of polymerisation,52,53 together with genetic factors,72,73 may account for the heterogeneity in liver disease among individuals who are homozygous for the Z mutation. The synthesis of AAT rises during episodes of inflammation as part of the acute phase response. At these times the formation of polymers is likely to overwhelm the degradative pathway, thereby exacerbating the formation of hepatic inclusions and the associated hepatocellular damage. This hypothesis has been challenged by cell studies which do not show an increase in intracellular Z AAT in response to raised temperatures.74 However, our recent data in a Drosophila model of AAT deficiency show a clear temperature dependence of polymerisation in vivo.75 There is also anecdotal clinical evidence to support the role of temperature in exacerbating the liver disease associated with Z AAT from the prospective study of Sveger and colleagues in Sweden.45,46 They screened 200 000 newborn babies and identified 120 Z homozygotes whom they have followed into late adolescence. Two of these patients developed progressive jaundice during the course of the study; in one this followed an acute appendicitis and in the other severe pneumonia. Other asymptomatic infants developed marked derangement of liver function tests in association with coryzal illnesses and eczema. Further prospective studies are required to assess whether pyrexial episodes occur more frequently and increase the burden of intrahepatic polymers in Z AAT homozygotes who develop liver disease compared with those individuals who remain asymptomatic.
Although many AAT deficiency variants have been described, only two mutants (other than the Z allele) have similarly been associated with plasma deficiency and hepatic inclusions: AAT Siiyama (53serine→phenylalanine) which is the commonest cause of AAT deficiency in Japan,76,77 and Mmalton (also known as Mnichinan78 and Mcagliari,79 deletion of phenylalanine at position 52) which is the most common cause of AAT deficiency in Sardinia. Both of these mutants destabilise and open β-sheet A (fig 2) to allow the formation of folding intermediates61,62 and loop-sheet polymers in vivo.80,81 Polymerisation also underlies the mild plasma deficiency of the S (Glu264Val) and I (Arg39Cys) variants of AAT.82,83 The point mutations that are responsible for these variants cause less disruption to β-sheet A than does the Z variant. Thus, the rates of polymer formation are much slower than that of Z AAT53 and this results in less retention of protein within hepatocytes, milder plasma deficiency, and the lack of a clinical phenotype. However, if a mild slowly polymerising I or S variant of AAT is inherited with a rapidly polymerising Z variant, the two can interact to form heteropolymers within hepatocytes leading to inclusions and finally cirrhosis.83–85
MOLECULAR PATHOLOGY OF THE LUNG DISEASE ASSOCIATED WITH PI Z AAT
The single most important factor in the development of emphysema in patients with AAT deficiency is smoking.49,86 The combination of antiproteinase deficiency and cigarette smoke can have a devastating effect on lung function,48,87 probably by allowing the unopposed action of proteolytic enzymes. AAT levels are greatly reduced in the lungs of individuals with AAT deficiency.88 Moreover, the AAT that is available to protect the lungs is approximately five times less effective at inhibiting neutrophil elastase than normal M AAT.55,89–91 The inhibitory activity of Z AAT can be further reduced as AAT is susceptible to inactivation by oxidation of the P1 methionine residue by free radicals from leucocytes or direct oxidation by cigarette smoke.5,6,92,93 Finally, the Z mutation favours the spontaneous formation of AAT loop-sheet polymers within the lung.94 This conformational transition inactivates AAT as a proteinase inhibitor, thereby further reducing the already depleted levels of AAT that are available to protect the alveoli (fig 4). The mechanisms that drive the formation of Z AAT polymers within the lung are unknown. It is possible that polymerisation may be accelerated by the inflammatory milieu that exists within the lungs of individuals with Z AAT deficiency. Moreover, cigarette smoke is mildly acidic and previous studies have shown that polymerisation of AAT is accelerated at low pH.53 Thus, cigarette smoke may act in several ways to promote the inactivation of Z AAT in vivo.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed model for the pathogenesis of emphysema in patients with Z α1-antitrypsin (AAT) deficiency. The plasma deficiency and reduced inhibitory activity of Z AAT may be exacerbated by the polymerisation of AAT within the lungs. These processes inactivate the inhibitor, thereby further reducing the antiproteinase screen. AAT polymers may also act as a proinflammatory stimulus to attract and activate neutrophils, thereby increasing tissue damage. Reproduced from Lomas and Mahadeva13 with permission.
Patients with Z AAT deficiency have an excess number of neutrophils in bronchoalveolar lavage fluid95 and in tissue sections of lung parenchyma13 compared with controls. This may reflect an excess of chemoattractant agents such as leukotriene B4 (LTB4) and interleukin (IL)-8.96,97 However, recent studies have shown that the polymers are themselves chemotactic for human neutrophils in vitro.98 The magnitude of this effect was similar to the chemoattractant C5a and was present over a range of physiological concentrations (EC50 4.5 (2) μg/ml). Polymers also induced neutrophil shape change and stimulated myeloperoxidase release and neutrophil adhesion.98 It is possible that polymers of Z AAT form in vivo and then act as a chronic chemoattractant to cause an influx of inflammatory cells.13 They may evade the defence mechanisms of the lung by adhering to the interstitium. Any proinflammatory effect of polymers is likely to be exacerbated by inflammatory cytokines, cleaved or complexed AAT,99 elastin degradation products,100 and cigarette smoke which themselves cause neutrophil recruitment. Our understanding of the biological properties of AAT thus provides novel pathways for the pathogenesis of emphysema in individuals who are homozygous for the Z mutation (fig 4). Indeed, the presence of polymers may explain the progression of lung disease in Z AAT homozygotes after smoking cessation despite adequate intravenous replacement with plasma AAT. The relationship between intrapulmonary Z AAT polymers and smoking, infection, cytokine production, and rate of decline in lung function requires assessment in both cell and animal models of disease, and prospective studies in Z homozygotes
MOLECULAR PATHOLOGY OF OTHER CONDITIONS ASSOCIATED WITH PI Z AAT
Pi Z AAT deficiency is reported with panniculitis that is characterised by an acute inflammatory infiltrate of the skin and fat necrosis.101,102 There is also an association of the Z allele of AAT with asthma,103,104 vasculitis,105,106 bronchiectasis,107 pancreatitis,108 and vascular aneurysms,109,110 although the association with bronchiectasis and vascular disease have been disputed by other studies.108,111 The feature that links many of these conditions is neutrophil mediated inflammation, and it is possible that AAT polymers are one of the factors that drive this inflammation and disease progression.98
DISEASE CAUSED BY THE POLYMERISATION OF OTHER SERPINS
AAT is the archetypal member of the serine proteinase inhibitor or serpin superfamily. This family includes members such as α1-antichymotrypsin, C1 inhibitor, antithrombin, and plasminogen activator inhibitor-1 which have an important role in the control of proteinases involved in the inflammatory, complement, coagulation, and fibrinolytic cascades, respectively.25,112 The family is characterised by more than 30% sequence homology with AAT and conservation of tertiary structure.15,113 Consequently, physiological and pathological processes that affect one member may be extrapolated to another. The phenomenon of loop-sheet polymerisation is not restricted to AAT and has now been reported in mutants of other members of the serpin superfamily to cause disease (the serpinopathies). Mutants of C1 inhibitor, antithrombin and α1-antichymotrypsin can also destabilise the protein architecture to form inactive polymers that are associated with plasma deficiency and angio-oedema, thrombosis, and chronic obstructive pulmonary disease, respectively.59,114–120
The process is most strikingly displayed by the inclusion body dementia, familial encephalopathy with neuroserpin inclusion bodies (FENIB).121 We have shown that this dementia is caused by mutations in neuroserpin that are homologous to those causing liver cirrhosis in AAT deficiency.121 Moreover, both the liver cirrhosis and the neurodegenerative disease have an identical pattern of intracellular polymerisation and inclusion body formation (fig 3). Further kindreds with polymerogenic neuroserpin mutations have been described and it is becoming clear that there is a direct relationship between the magnitude of intracellular accumulation of neuroserpin and the severity of the clinical syndrome.122 Moreover, our recent work has shown that one of the neuroserpin mutants that causes FENIB (Ser49Pro) polymerises up to 13-fold faster than wild type protein.123 This provides strong support for the role of aberrant neuroserpin polymerisation in the pathogenesis of FENIB.
PREVENTION OF POLYMER FORMATION
There is substantial evidence that polymers of AAT and, indeed, of all other serpins form by an aberrant linkage between the reactive centre loop of one molecule and β-sheet A of another.26,52,54,124–127 This has allowed the development of new strategies to attenuate polymerisation and so treat the associated disease. We have shown previously that the polymerisation of Z AAT can be blocked by annealing of reactive loop peptides to β-sheet A.52,128 Such peptides were 11–13 residues in length and could bind to other members of the serpin superfamily.128,129 This was most clearly demonstrated by the finding that the reactive loop peptide of antithrombin inserted more readily to β-sheet A of AAT and vice versa.130 These peptides, although useful in establishing the mechanism of polymerisation, are too long and too promiscuous to be suitable for rational drug design. More recently we have designed a 6-mer peptide that specifically anneals to Z AAT alone and blocks polymerisation.63 Indeed, trimer peptides have been developed that will also anneal to a patent β-sheet A of antithrombin in vitro.131 The aim now is to convert these peptides into small drugs that can be used in vivo.
A second strategy comes from the identification of a hydrophobic pocket in AAT that is bounded by strand 2A and helices D and E.29,132 The cavity is patent in the native protein but is filled as β-sheet A accepts an exogenous reactive loop peptide during polymerisation.29 We have shown that introducing mutations into this pocket retards the polymerisation of M AAT and increases the secretion of Z AAT from a Xenopus oocyte expression system.133 This cavity is therefore an ideal target for the development of drugs that will stabilise β-sheet A and so ameliorate polymer formation.
An alternative strategy is to use chemical chaperones to stabilise intermediates on the folding pathway. Osmolytes such as betaine, trimethylamine oxide, and sarcosine all stabilise AAT against polymer formation.134 The chaperone trimethyamine oxide had no effect on the secretion of Z AAT in cell culture74 as it favoured the conversion of unfolded Z AAT to polymers.135 In contrast, glycerol increased the secretion of Z AAT from cell lines74 most likely as it binds to and stabilises β-sheet A.131 4-phenylbutyrate (4-PBA) also increased the secretion of Z AAT from cell lines and transgenic mice.74 This agent has been used for several years to treat children with urea cycle disorders and, more recently, 4-PBA has been shown to increase the expression of mutant (ΔF508) cystic fibrosis transmembrane regulator protein in vitro136 and in vivo.137 These encouraging findings have led to a pilot study that is currently ongoing to evaluate the potential of 4-PBA to promote the secretion of AAT in patients with AAT deficiency.
CONCLUSION
The molecular basis of Z AAT deficiency has now been elucidated with biochemical, cellular, and structural studies. The current goals are to determine the cellular response to polymeric AAT and to develop therapeutic strategies to block polymerisation in vivo.
Acknowledgments
This work is supported by the Medical Research Council (UK), the Wellcome Trust, the Alpha-one Foundation and Papworth NHS trust. HP is an MRC Training Fellow and the recipient of a Sackler Fellowship.