Article Text

Abstract
Background: In patients with cystic fibrosis (CF) neutrophils are recruited in excess to the airways yet pathogens are not cleared and the patients suffer from chronic infections. Recent studies have shown a deficiency in airway fluids from patients with CF and other inflammatory pulmonary conditions of surfactant protein A (SP-A), a pattern recognition molecule that facilitates uptake of microbes by macrophages and neutrophils.
Methods: In vitro simulations were used to test the hypothesis that decreased SP-A levels in CF might be the result of degradation by neutrophil serine proteases.
Results: Very low levels of the neutrophil granule serine proteases cathepsin G, elastase, and proteinase-3 rapidly degraded pure SP-A when tested in buffered saline. The order of potency was cathepsin G>elastase>proteinase-3. The addition of cathepsin G or elastase to normal bronchoalveolar lavage (BAL) fluid caused a dose dependent degradation of endogenous native SP-A. Cathepsin G and elastase were present in the BAL fluid from many patients with CF. Simple incubation of protease positive BAL fluid from patients with CF caused a time dependent degradation of added SP-A or, where present, endogenous SP-A. The degradation of SP-A by protease(s) in BAL fluid of patients with CF was abrogated by diisopropylfluorophosphate and monocyte/neutrophil elastase inhibitor.
Conclusions: The findings strongly suggest that the neutrophil serine proteases cathepsin G and/or elastase and/or proteinase-3 contribute to degradation of SP-A and thereby diminish innate pulmonary antimicrobial defence.
- inflammation
- neutrophil serine proteases
- cathepsin G
- surfactant protein A
- cystic fibrosis
- BAL, bronchoalveolar lavage
- CF, cystic fibrosis
- CRD, carbohydrate recognition domain
- DFP, diisopropyl fluorophosphate
- MeOSuc, methoxysuccinyl
- MNEI, monocyte/neutrophil elastase inhibitor
- PVDF, polyvinyldine difluoride
- SBzl, thiobenzyl ester
- SP-A, surfactant protein-A
- Suc, succinyl
Statistics from Altmetric.com
- BAL, bronchoalveolar lavage
- CF, cystic fibrosis
- CRD, carbohydrate recognition domain
- DFP, diisopropyl fluorophosphate
- MeOSuc, methoxysuccinyl
- MNEI, monocyte/neutrophil elastase inhibitor
- PVDF, polyvinyldine difluoride
- SBzl, thiobenzyl ester
- SP-A, surfactant protein-A
- Suc, succinyl
There is increasing evidence to suggest that surfactant protein-A (SP-A) is a cofactor with macrophages in pulmonary antimicrobial and anti-inflammatory defence.1 SP-A is an abundant hydrophilic protein associated with surfactant phospholipids lining the airways. It is a multimeric collectin protein (collagenous lectin). Mice depleted of SP-A have decreased clearance of intratracheally delivered pathogens including group B streptococci, Pseudomonas aeruginosa, and respiratory syncytial virus, coincident with decreased uptake by macrophages and enhanced neutrophil infiltration and synthesis of inflammatory cytokines.2 In vitro, SP-A enhances phagocytosis of pathogens by macrophages and neutrophils dependent in part on its function as a “pattern recognition molecule”3 that binds, generally via the lectin domain, to pathogen associated molecular patterns and thereby opsonises the microorganisms. SP-A also acts directly on macrophages to enhance phagocytic capacity and modulate cytokine production.4
Decreased SP-A levels have been found in several respiratory diseases including bacterial pneumonia, adult respiratory distress syndrome,5,6 and cystic fibrosis.7,8 Common features are the deterioration of pulmonary antimicrobial defence and the presence of inflammation.
Cumulative evidence suggests that decreased SP-A levels in some of these disease states result from degradation by neutrophil proteases. Prime candidates for this putative pathological action are the related serine proteases elastase, cathepsin G and proteinase-3 carried in human neutrophil azurophil granules at very high levels (1.1 pg elastase, 0.8 pg cathepsin G per neutrophil).9 To explore this postulate we examined the neutrophil serine proteases for their capacity to degrade SP-A in a pure protein format and as an endogenous protein in bronchoalveolar lavage (BAL) fluid.
We also studied BAL fluid from patients with cystic fibrosis (CF), an extreme natural model of neutrophil dominated airway inflammation that includes deficient antimicrobial defence and decreased SP-A levels. In CF, inherited defects in CFTR, an apical surface membrane protein of epithelial cells, lead to progressive pulmonary disease which accounts for most of the morbidity and mortality.10 CF lung disease is a continuous process of infection and inflammation.11 Inflammatory markers, particularly interleukin (IL)-8, are found in airway fluids of patients with CF.12 Elastase released from neutrophils into the airway epithelial lining fluid of CF patients forms a complex with protease inhibitors and, when the protease inhibitor shield is overwhelmed, is detected as active protease.13 Whereas no free elastase was found in the airway epithelial lining fluid of healthy subjects, mean levels of 8.2 μM (~200 μg/ml) were found in patients with CF with moderate lung function impairment14 and 2.3 μM (~60 μg/ml) in those with mild disease.15 In the present study we tested BAL fluid samples from patients with CF for the presence of SP-A degrading activity and for cathepsin G and elastase content.
METHODS
Materials
SP-A isolated from lung lavage fluid of patients with alveolar proteinosis was obtained from Chemicon International (Temecula, CA, USA) and stored at 1 mg/ml in 10× phosphate buffered saline (PBS) in aliquots at −20°C. Human neutrophil elastase purified from purulent human sputum was obtained from Elastin Products (Owensville, MO, USA); human neutrophil cathepsin G and proteinase-3 isolated from blood leucocytes were from Athens Research and Technology (Athens, GA, USA). Proteases were stored in aliquots at 1 mg/ml at −20°C in PBS (neutrophil elastase and cathepsin-G) or 50 mM MES, pH 4.5, 0.7 M NaCl (proteinase-3). Recombinant human monocyte/neutrophil elastase inhibitor (MNEI)16 was produced in insect cells, purified, and stored in aliquots in PBS at 2.3 mg/ml at −80°C. Diisopropyl fluorophosphate (DFP) was obtained from Sigma Chemical Co (St Louis, MO, USA).
BAL fluid from CF and non-CF control patients
BAL fluid samples were obtained as part of a cross sectional study of airway inflammation and infection in CF approved by the University of Colorado Institutional Review Board.12 Bronchoscopy and bronchoalveolar lavage were performed at The Children’s Hospital, Pediatric Pulmonary Medicine, Denver, CO, USA. Patients were sedated with midazolam (0.1–0.3 mg/kg) and fentanyl (1–4 μg/kg) and topical lidocaine was applied to the airway. Three consecutive washes of sterile saline at room temperature (total volume 3 ml/kg for infants and 80 ml for older patients) were instilled through a bronchoscope into the lingula or right middle lobe. BAL fluid was aspirated immediately by gentle suction and pooled. Average lavage fluid return was 50%.
Two to four ml of lavage fluid was set aside for total and differential cell counts, cytopathology, and viral and bacterial culture, and the remainder was centrifuged at 1200×g for 10 minutes. The cell free supernatant (BAL fluid) was immediately frozen in aliquots at −80°C. Core parameters of inflammation were measured for all BAL fluid samples; in addition to neutrophil count, these included free elastase measured by hydrolysis of MeO-Suc-Ala-Ala-Pro-Ala-p-nitroanilide (Sigma) at 410 nm. The inhibitor MeO-Suc-Ala-Ala-Pro-Val-CH2Cl (Sigma) was used to confirm the specificity of the elastase activity.
For the present study of SP-A degradation, aliquots of BAL fluid from16 patients with CF (11 elastase positive and five elastase negative) were randomly drawn. Sixteen elastase negative control (non-CF) aliquots of BAL fluid were also randomly drawn from patients with chronic cough (n = 3), asthma (n = 3), interstitial lung disease (n = 3), immunodeficiency (n = 2), and one each with pulmonary haemosiderosis, hypoxemia, chronic aspiration, suspected aspiration, and haemorrhage.
The median age of the subjects with CF was 9.4 years (range 1.4–23.3) and 9.2 years (range 1.2–19.3) for the non-CF patients. BAL fluid urea levels were similar in both groups (mean 0.55 mg/dl (range 0.20–1.10) for the CF patients and 0.46 mg/dl (0.1–1.40) for the non-CF patients), indicating that BAL was performed comparably in the two groups.
BAL fluid samples from four of the non-CF patients and four of the CF patients were studied for cleavage of SP-A in vitro (coded identifiers appear in the legends of figs 3 and 4). The major criterion for sample selection was the availability of sufficient volume of the specimen for study; other criteria are shown in the accompanying text and figure legends.
Quantitation of active cathepsin G
Cathepsin G activity was quantified by a coupled spectrophotometric reaction. BAL fluid (50 μl) or BAL fluid diluted in PBS with 0.05% Tween-20 (PBS-Tween) was incubated in 1 ml of 20 mM Tris-HCl, pH 7.4, 500 mM NaCl, 0.1% polyethylene glycol with 0.2 mM of the thiobenzyl (SBzl) substrate Suc-Ala-Ala-Pro-Phe-SBzl (Bachem, King of Prussia, PA, USA) and 0.5 mM Ellman’s reagent (5,5′-dithiobis(2-nitrobenzoic acid; Sigma) for 5 minutes at ~22°C. OD412 nM was recorded at 15 second intervals. Pure neutrophil cathepsin G was used to generate reference curves. Preliminary experiments showed that the response was linear from 0.1 μg/ml (the threshold level) to 1.0 μg/ml. Neutrophil elastase at 20 μg/ml and proteinase-3 at 10 μg/ml were without activity. The coefficient of variance was 10%. For specific inhibition, BAL fluid samples were preincubated for 5 minutes with MNEI which inhibited the activity of the samples by ⩾90%.
Proteolysis reactions
Pure SP-A (1 μg) was incubated with 3–100 ng elastase, cathepsin G, or proteinase-3 in 18 μl PBS-Tween at 37°C for 10 minutes. BAL fluid samples (15 μl) were combined with protease or SP-A in a final volume of 18 μl and incubated at 37°C for varying time periods. Where indicated, protease inhibitors (DFP or MNEI) were added before incubation. The reactions were stopped by adding DFP (2 mM), incubating at ~22°C for 3 minutes, adding 5× solubilising solution (10% SDS, 25% glycerol, 0.1 mg/ml bromphenol blue, 5% β-mercaptoethanol), and heating at 100°C for 2 minutes. The samples were electrophoresed on 10% or 4–20% acrylamide gels; Novex, San Diego, CA, USA). The products from the BAL fluid experiments were transferred to nitrocellulose for antibody staining (described below). The products of pure SP-A were stained with silver (Silver Stain Plus, Bio-Rad, Hercules, CA, USA) or electrophoretically transferred to polyvinylidene fluoride (PVDF; Millipore Corp, Bedford, MA, USA) for gold staining (Aurodye, Amersham Pharmacia Biotech, Piscataway, NJ, USA). Because SP-A reacts only weakly with protein stains (preliminary experiments, data not shown), the more sensitive gold stain was used for most pure protein experiments.
Western blot analysis for SP-A
The nitrocellulose transfers were blocked with 20% milk solids in PBS-Tween at ~22°C and stained for 1 hour with PE-10 or PC-6 mouse monoclonal antibodies to human SP-A17 provided by Dr Yoshio Kuroki (Sapporo Medical University, Sapporo, Japan) (0.6 μg/ml monoclonal antibody in PBS-Tween-0.1% milk solids). PE-10 and PC-6 were reported to bind within the carbohydrate recognition domain (CRD), although reports of exact epitope locations are conflicting.17–19 The blots were washed with PBS-Tween and incubated with 125I-labelled rabbit anti-mouse IgG. Reactive SP-A bands at ~34 kDa were captured with the Storm 860 phosphorimager (Molecular Dynamics, Sunnyvale, CA, USA) and quantified using ImageQuant version 1.2 software (Molecular Dynamics) by comparison with dilutions of pure SP-A electrophoresed in parallel. SP-A levels for patient BAL fluid samples are the mean of triplicate assays; the lower limit of linearity was 0.6 μg/ml BAL fluid and the coefficient of variance was 6%.
RESULTS
Degradation of pure SP-A by neutrophil elastase, cathepsin G, and proteinase-3
As the first approach to determining the effect of the neutrophil serine proteases, pure SP-A (from patients with alveolar proteinosis) was incubated in buffered saline with varying amounts of elastase, cathepsin G, or proteinase-3. The reaction products were analysed by SDS-PAGE. Gold stained transfers showed that each of the proteases degrades SP-A in a dose dependent manner (fig 1A). Cathepsin G, a protease with chymotrypsin-like specificity,20 was most potent with only 1 μg/ml (0.04 μM) causing 50% degradation of SP-A (60 μg/ml) in 10 minutes, followed by elastase and proteinase-3 for which 3 μg/ml (0.12 μM) and an estimated 10 μg/ml (0.40 μM), respectively, were required for 50% cleavage (fig 1B).

Degradation of SP-A by elastase (ela), cathepsin G (cat G), and proteinase-3 (pr-3). (A) SP-A (60 μg/ml) was incubated with the indicated protease concentrations in PBS-Tween at 37°C for 10 minutes. The products were resolved by SDS-PAGE (reducing conditions), transferred to PVDF, and stained with AuroDye (gold stain). Note that SP-A, a monomer of 34 kDa (SP-A) and a dimer of 68 kDa, is degraded by each protease and that cathepsin G is most effective. No SP-A fragment was detected, even on 4–20% gradient gels (not shown); the 26 kDa bands in the right lanes are the proteases. Silver stained gels also revealed loss of SP-A but did not show fragments (data not shown). (B) Mean (SE) data for the three experiments.
No SP-A fragments were detected after gold or silver staining of 10% or 4–20% acrylamide gels (~7 kDa cut-off; fig 1), suggesting that the initial SP-A fragments are short lived. To confirm that SP-A loss is due to proteolysis, the incubations were carried out in the presence of protease inhibitors. Loss of SP-A was abrogated by DFP, a 200 dalton broad spectrum inhibitor of serine proteases21 (fig 2A) and MNEI, a 42 kDa serpin inhibitor of elastase-like proteases and chymotrypsin-like proteases including cathepsin G16 (fig 2B), indicating that cathepsin G and elastase mediated loss of SP-A is indeed due to proteolytic degradation. Inhibition of SP-A proteolysis by MNEI was partial when the inhibitor:protease (I:P) ratio was less than 1 and complete when the I:P ratio was greater than 1 (fig 2B), consistent with the stoichiometric inhibition mechanism of serpins. The association rate constants for the reactions of MNEI with elastase, cathepsin G, and proteinase-3 were 3.4×107/mol/s, 2.3×106/mol/s, and 1.7×107/mol/s, respectively.16

Cathepsin G and elastase mediated loss of SP-A is abrogated by protease inhibitors. Elastase and cathepsin G were preincubated (3 minutes at ~22°C) without or with (A) 2 mM diisopropyl fluorophosphate (DFP) or (B) monocyte/neutrophil elastase inhibitor (MNEI) at the indicated inhibitor:protease molar ratio. SP-A was added and the reactions (60 μg/ml SP-A, 5 μg/ml protease) were incubated for 10 minutes. Gold stained transfers from a representative experiment out of three are shown.
Degradation of endogenous SP-A in control BAL fluid by added elastase and cathepsin G
To study SP-A in an approximation of the pulmonary environment we exposed minimally manipulated BAL fluid samples from control patients to elastase and cathepsin G. Because processing of BAL fluid is done rapidly and involves only low speed centrifugation, the endogenous SP-A molecules in the BAL fluid probably retain native conformation and interactions. Elastase and cathepsin G were added and the reactants were incubated for 10 minutes and analysed by Western blot analysis. (The high protein content of BAL fluid precluded protein staining.) The level of SP-A was decreased by each protease in a dose dependent manner (fig 3A) but was unchanged when the BAL fluid was incubated without added protease (fig 3B, lane 2). These findings indicate that cathepsin G and elastase degrade native conformation SP-A in the presence of airway fluid components.

Degradation of endogenous SP-A in BAL fluid by added cathepsin G and elastase. (A) BAL fluid from a control patient (N-10) was incubated for 10 minutes with cathepsin G or elastase. Western blots stained for PE-10 antibody are shown. (B) Time course of SP-A degradation in control BAL fluid incubated with 3 μg/ml cathepsin G. The PE-10 stained blot is from a 4–20% gel. The SP-A fragment (~20 kDa) was also found in elastase treated BAL fluid at trace levels and was detected with PC-6 (not shown). (C) Quantitation of SP-A degradation in four control BAL fluid samples. Mean (SE) levels of SP-A are shown as a percentage of the starting amount. The starting SP-A content for BAL fluid samples N-06, N-08, N-09 and N-10 were 5.2, 10.7, 7.9 and 8.3 μg/ml, respectively (mean of duplicate assays). Note that cathepsin G was more potent than elastase in degrading endogenous SP-A.
Trace levels of a ~20 kDa SP-A fragment were found in PE-10 and PC-6 stained blots of BAL fluid treated with cathepsin G and elastase (fig 3B). Based on epitope specificity of the two antibodies, this fragment is likely to include regions of the CRD. When examined as a function of time, the amount of ~20 kDa fragment was greatest at the earliest time points (1 and 3 minutes) but was substantially less than the amount of SP-A lost, suggesting that the cleavage product is more sensitive to proteolysis than intact SP-A.
Quantitation of endogenous SP-A degradation for four control BAL fluid samples showed that cathepsin G was more potent than elastase in degrading SP-A (fig 3C), strongly suggesting that cathepsin G may also be more potent in vivo. For each of the four BAL fluid samples, low levels of the proteases were sufficient for complete or substantial degradation of endogenous SP-A in 10 minutes.
Cathepsin G and elastase in BAL fluid of CF patients
The level of free elastase in BAL fluid specimens was measured at the time of sample collection by hydrolysis of MeO-Suc-Ala-Ala-Pro-Ala-p-nitroanilide. Because of the effective SP-A cleaving activity of cathepsin G, we set up an assay for this protease (hydrolysis of Suc-Ala-Ala-Pro-Phe-thiobenzyl ester). Sixteen randomly drawn BAL fluid samples from patients with CF (CF101-CF116) and 16 control (non-CF) subjects (N01–N16) were assayed. Active cathepsin G was found in all of the 11 elastase positive BAL fluid samples from the CF patients, in one of the five elastase negative BAL fluid samples from CF patients, and in one of 16 elastase negative control BAL fluid samples (table 1). The remaining four CF BAL fluid samples and 15 control samples lacked cathepsin G. The levels of cathepsin G in BAL fluid from patients with CF varied over a large range, the highest being more than 19 μg/ml. Although their ratios varied, the levels of elastase and cathepsin G were highly correlated (r2 = 0.89), strongly suggesting a common origin.
Levels of cathepsin G and elastase in BAL fluid from control patients (N01–N16) and patients with CF (CF101–116)
Degradation of SP-A by serine proteases in BAL fluid of CF patients
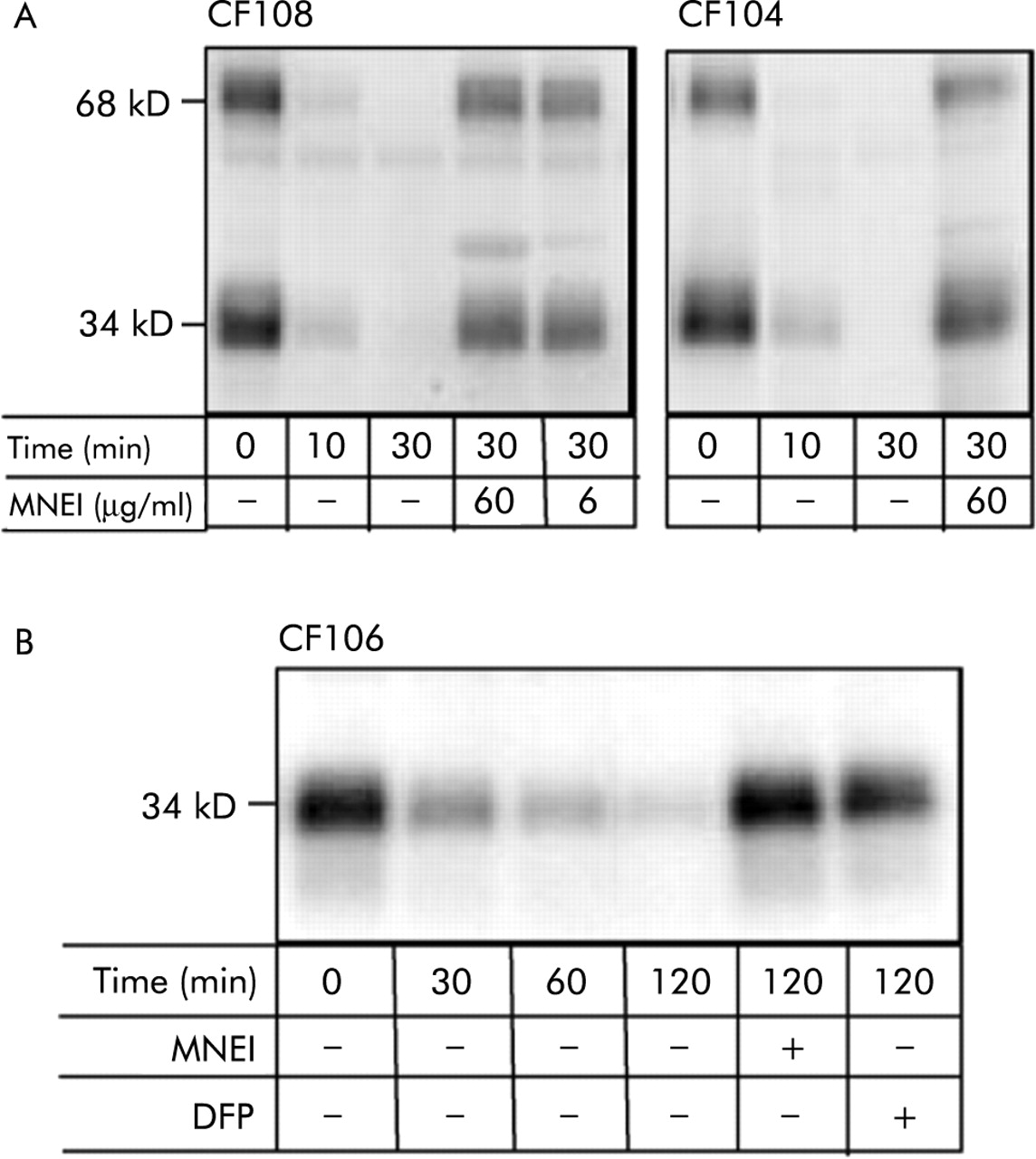
To determine whether SP-A is degraded by proteases in inflamed airways of patients with CF, BAL fluid samples from patients CF108 and CF104 which contained active elastase and cathepsin G were examined. These BAL fluid samples had no detectable SP-A (data not shown) so pure SP-A (5 μg/ml) was added. On incubation, time dependent disappearance of SP-A was observed that was almost complete by 30 minutes (fig 4A). The degradation of added SP-A by endogenous protease(s) in BAL fluid from CF patients was abrogated by DFP (not shown) and MNEI (fig 4A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Degradation of SP-A by serine protease(s) in BAL fluid of patients with CF. (A) Degradation of added SP-A. BAL fluid samples CF108 (left) and CF104 (right) contain active elastase and cathepsin G (table 1) and have no detectable SP-A. Pure SP-A (5 μg/ml) was added and the mixtures incubated at 37°C for the indicated time. PE-10 stained Western blots are shown. Note that added SP-A was degraded by endogenous protease. Diisopropyl fluorophosphate (DFP) (not shown) and monocyte/neutrophil elastase inhibitor (MNEI) prevented the reaction. (B) Cleavage of endogenous SP-A. BAL fluid sample CF106, which contains SP-A (5.8 μg/ml) as well as active proteases, was incubated without additive at 37°C. Note that endogenous SP-A was degraded over 2 hours by endogenous protease and that MNEI and DFP abrogated degradation. Similar results were found for BAL fluid sample CF110 (2.1 μg SP-A/ml) except that 3 hours were required to degrade SP-A (not shown). The results are representative of duplicate experiments.
Two additional BAL fluid samples, CF 106 and CF110, were also studied. These contained endogenous SP-A in addition to elastase and cathepsin G. When these specimens were incubated without additive, time dependent degradation of endogenous SP-A occurred over 2–3 hours (fig 4B). Degradation of endogenous SP-A by endogenous protease in BAL fluid from patients with CF was also abrogated by DFP and MNEI (fig 4B).
DISCUSSION
The findings of this study provide substantial in vitro evidence indicating that active neutrophil serine proteases present in inflammatory airway fluids cleave SP-A and thereby negatively impact a host of SP-A mediated innate antimicrobial and anti-inflammatory functions. When tested in a pure protein format, each of the neutrophil azurophil granule proteases rapidly degraded pure SP-A with the rank order: cathepsin G>elastase>proteinase-3. Low levels of the two tested proteases, cathepsin G and elastase, also rapidly degraded endogenous SP-A in minimally manipulated non-CF BAL fluid samples. In the third protocol proteases present in inflammatory airway fluids of patients with CF served as SP-A cleaving enzymes. On simple incubation, residual endogenous SP-A or pure added SP-A was degraded by endogenous inflammatory proteases within several hours, which suggests that proteases are important in loss of SP-A. Abrogation of in vitro degradation by DFP identified the SP-A cleaving protease in BAL fluid from CF patients as a serine active site enzyme(s), and abrogation by MNEI strongly implicated neutrophil elastase and/or cathepsin G and/or proteinase-3.
The levels of cathepsin G and elastase required for complete degradation of SP-A are well within levels documented for CF patients, even for patients with mild impairment of lung function who have ~60 μg/ml elastase in airway epithelial lining fluid15 (see table 1 for patient BAL fluid data which are approximately 30-fold diluted relative to epithelial lining fluid). Cathepsin G at 1 μg/ml and elastase at 3 μg/ml degraded 50% of the starting levels of SP-A (60 μg/ml) in incubations that were limited to 10 minutes. Similar levels of cathepsin G and elastase degraded ~50% of the starting SP-A in BAL fluid (6–10 μg/ml) in the presence of various airway fluid components. The duration of the latter reactions was also limited to 10 minutes. The use of control BAL fluid as a source of the target protein showed that cathepsin G and elastase degrade SP-A in the presence of airway fluid components. Because SP-A in minimally manipulated BAL fluid probably retains its native conformation and association with protein and lipid components, the use of this material as an experimental system strengthens the evidence for an enzyme:target relationship.
Although not as well studied as elastase, cathepsin G22,23 and proteinase-324 have also been found in airway fluids of patients with CF. One study of CF sputum found nearly equal levels of cathepsin G and elastase,23 as was anticipated because of the nearly equal content of the two enzymes in neutrophils (1.1 pg elastase per cell, 0.8 pg cathepsin G)9 and their shared mechanisms of release. Schuster et al22 found lower levels of cathepsin G than elastase in sputum, similar to our findings in BAL fluid. Because only activity was measured, the lower cathepsin G level might reflect its specific inhibition. Also, although the three neutrophil granule serine proteases are cationic, the rank order is cathepsin G>elastase>proteinase-3, so cathepsin G might bind preferentially to negative surfaces such as cell surfaces,25 mucus, or DNA23,26 and not be harvested in cell-free BAL fluid.
The postulate that inflammatory neutrophil protease, specifically elastase, destroys SP-A has been previously suggested and was discussed within the context of CF in a recent review.27 Earlier studies have shown that canine and bovine SP-A are degraded by elastase and by supernatants of activated neutrophils.28–30 The present study extends these findings to human SP-A and BAL fluid and shows that the degrading activity of SP-A is a capacity shared by all three neutrophil granule serine proteases.
Using a proteomics approach, von Bredow et al found small SP-A degradation fragments in BAL fluid from CF but not from non-CF patients.31 Following 8 weeks of aerosol treatment with the serine proteinase inhibitor α1-antitrypsin (α1-protease inhibitor), SP-A fragments decreased, suggesting that proteases contribute to SP-A degradation in CF and that antiprotease treatment may decrease degradation.32
Cumulatively, the present and previous studies suggest that neutrophil serine proteases, particularly cathepsin G and elastase, are pathological agents that destroy SP-A in CF airways and thereby contribute to loss of innate pulmonary antimicrobial defence. Whether a similar situation occurs in non-CF respiratory conditions with decreased SP-A levels such as bacterial and viral pneumonia, acute respiratory distress syndrome, and idiopathic pulmonary fibrosis5,6,33 is at present more speculative. The inflammation component is well documented and, although elastase activity is generally not detected in BAL fluid, this does not mean that SP-A in patient airways is not exposed to neutrophil serine proteases. These enzymes, when released into extracellular space, are expected to act locally and transiently, with their activity terminated when diffusion permits encounter with protease inhibitors.34 For elastase or cathepsin G to be detected enzymatically in harvested BAL fluid, the amount must be sufficient to overwhelm the total protease inhibitor shield of the airway. Thus, enzyme assay detects only the most extreme cases of in vivo exposure to neutrophil proteases which are frequent in CF. Further studies are needed to determine whether an excess of neutrophil proteases sufficient to degrade SP-A is transiently present in non-CF patients with inflammatory conditions. Possible evidence would include a decrease in the elastase inhibitory activity of BAL fluid or the presence of elastase bound to protease inhibitors.
We recognise that a detailed analysis of BAL fluid was performed on a limited number of specimens from CF patients and does not rule out the possibility of contributions from other proteases or other mechanisms to the pathological decrease in SP-A. Indeed, a recent study showed that SP-A is degraded by a metalloprotease produced by Pseudomonas aeruginosa.35 That endogenous SP-A was degraded by endogenous proteases in vitro may have resulted from the mixing of fluid from the alveolar and airway compartments in the lavage fluid. It is likely that SP-A in the airway would be subject to an enormous burden of proteases. In our small series there was no statistically significant correlation between SP-A and either cathepsin G or neutrophil elastase, although samples with the highest cathepsin G and neutrophil elastase levels had low levels of SP-A. In a larger series, inverse correlations were seen in CF patients with airway infection and high neutrophil counts.36
The cumulative evidence suggests a causal link in respiratory inflammation, particularly in CF, of degradation of SP-A mediated by neutrophil serine protease and the loss of antimicrobial innate protection. The finding that each of the neutrophil azurophil granule serine proteases degrades SP-A emphasises the importance of considering these enzymes collectively as a source of destruction in inflammatory conditions. Drugs that target all three destructive proteases may be required to prevent loss of antimicrobial activity.
Acknowledgments
The authors thank Dr Y Kuroki for kindly providing antibodies, Dr G Caughey for valuable discussion, Drs C Benarafa, S Das and H Remold for critical reading of the manuscript and K Ronaszeki, W Nolin, I Osberg and M Sontag for technical contributions.
REFERENCES
Footnotes
-
Funding: This work was supported by the Cystic Fibrosis Foundation, Mike McMorris Cystic Fibrosis Center, and NIH grants HL66548 and RR00069 General Clinical Research Centers Program of the NCRR as well as a student fellowship award to FR from the Office of Enrichment Programs at Harvard Medical School.