Article Text

Abstract
Background: Functional polymorphisms in the genes encoding superoxide dismutases (SOD)—that is, superoxide scavenging antioxidant enzymes—may play an important role in the development of inflammatory airway diseases such as asthma.
Methods: The allele frequencies of two missense polymorphisms of SOD genes (Ala16Val in MnSOD (SOD2) and Arg213Gly in ECSOD (SOD3)) were investigated in Finnish patients with asthma and compared with family based controls. Both variants have been shown to be functionally interesting in the lung. The polymorphism at the exon–intron 3 boundary of a third SOD, CuZnSOD (SOD1), was also included in the analysis.
Results: None of the SOD genetic variants studied appeared to be major genetic regulators in the development of asthma. We could exclude all models of inheritance that increased the risk of asthma more than 1.2 fold for MnSOD*Val (frequency of allele 0.74 in the population) and more than 6.6 fold for ECSOD*Gly213 (frequency of allele 0.03 in the population) compared with non-carriers. For the intronic polymorphism in CuZnSOD, a relative risk of more than 3.3 (frequency of allele 0.10 in the population) could be excluded.
Conclusions: It is highly unlikely that the functionally important genetic variants Ala16Val and Arg213Gly of SODs play a major role in the genetic susceptibility of asthma.
- asthma
- polymorphism
- genetics
- superoxide dismutase
Statistics from Altmetric.com
Enhanced oxidative stress has been implicated in the development of inflammatory diseases such as asthma.1 It can therefore be hypothesised that the genetic variability of antioxidant enzymes may play a role, not only in the primary oxidant resistance of the airways, but also in the development of asthma and the progression of the disease. Superoxide dismutases (SOD) are the only enzymes that convert superoxide radicals to hydrogen peroxide. The genes encoding these enzymes are located in different chromosomes and in all of them polymorphisms have been described. Copper-zinc SOD (CuZnSOD, SOD1) is encoded on 21q22.1 (OMIM#147450),2 manganese SOD (MnSOD, SOD2) on 6q25.3 (OMIM#147460),2 and extracellular SOD (ECSOD, SOD3) on 4p16.3–q21 (OMIM#185490).2 All these enzymes are expressed in human bronchial epithelium and alveolar macrophages.3–5 In addition, ECSOD is highly expressed in the pulmonary vasculature and interstitium.5
Mice lacking CuZnSOD are healthy and display no increased sensitivity to hyperoxia,6 whereas studies with transgenic CuZnSOD mice have shown that increased levels of CuZnSOD reduce pulmonary oxygen toxicity.7 In CuZnSOD at least 60 point mutations, including a potential single nucleotide polymorphism located in the exon–intron 3 boundary, have been linked to motor neurone degeneration (ALS1 locus, OMIM#105400).2,8 Functionally important common variants of the gene have not been described. The allele frequency of this polymorphism in the Finnish population is 0.1.9
MnSOD has been considered to be of critical importance in the antioxidant defence of the lung. MnSOD deficiency in mice is lethal,10,11 while transgenic mice are resistant to hyperoxia induced toxicity. MnSOD is synthesised in the cytoplasm as a precursor molecule containing a leader signal that is later removed during the transport of the molecule to the mitochondria.12,13 Single nucleotide polymorphism Ala16Val13,14 is common (Val allele frequency approximately 48%),15 and it has been suggested that it may change the secondary structure and mitochondrial targeting of the protein. MnSOD*Ala16 has been associated with breast cancer13,16 and sporadic motor neurone disease,17 whereas MnSOD*Val16 homozygosity has been linked to idiopathic cardiomyopathy18 and lung cancer.15
ECSOD deficient mice develop normally and are healthy but, when exposed to high oxygen tension, they develop severe lung damage.19 Transgenic mice overexpressing ECSOD have decreased lung toxicity in hyperoxia.20 Single nucleotide polymorphism causing an amino acid change from Arg to Gly at position 213 is located in the heparin binding domain of the gene.21 This polymorphism is rare, occurring in 4–6% of various populations.22,23 It leads to increased circulating ECSOD activity and decreased anchoring of ECSOD to heparin in the interstitium.24 This polymorphism has been described in three patients with chronic obstructive lung disease.8,25
Studies have suggested that SOD activity is decreased in asthmatic airways,26 but there are no studies on the polymorphisms of SODs in asthma. We have therefore performed an association study on the known polymorphisms in Finnish patients with asthma and their family based controls. Special emphasis was placed on MnSOD because of its potential significance as the most important SOD in the mammalian lung.11
METHODS
Subjects
The study population consisted of 149 nuclear families, including 179 asthma patients and their 267 non-affected family members. The families represent a random subset of 253 families recruited from Central Eastern Finland (Kainuu province) in two phases (December 1994 and 1996). Initially the probands were identified as having self-reported doctor diagnosed asthma. All patients had been interviewed by a doctor and all participants had completed a questionnaire about their respiratory symptoms and allergies. In addition, their disease history and the results of the diagnostic tests (spirometry, histamine or methacholine challenge test, expiratory peak flow measurements) were verified retrospectively.27–29 Eighty seven percent of these self-reported asthma patients were accepted as verified cases. The age range of the asthmatic subjects was 2–80 years (mean 40.3) and of the unaffected family members was 2–83 years (mean 42.3). The female to male ratio in the asthma and control groups was 2.5:1 and 1:1.3, respectively. Both the probands whose diagnosis could not be verified and the healthy family members who nevertheless reported asthma-like symptoms were classified as having an unknown phenotype.
The history of the population of Kainuu has been studied in depth. The region was settled during the 16th century by a few hundred Finnish founder families. During 25 generations the population has expanded to 60 000 inhabitants.30
The study was approved by the ethical committees of the Kainuu Central Hospital and Department of Medical Genetics, University of Helsinki.
SNP genotyping by specific restriction enzyme digestion sites
DNA was extracted from peripheral blood leucocytes by a standard non-enzymatic method. The following primer pairs were used for amplification: 5′CTATCCAGAAAACACGGTGGGCC3′ and 5′TCTATATTCAATCAAATGCTACAAAACC3′ for non-coding A>C substitution in CuZnSOD (at position +35 from exon3/intron 3 boundary), 5′AGCCCAGCCGTGCGTAGAC3′ and 5′TACTTCTCCTCGGTGACG3′ for Val16Ala in MnSOD, and 5′CGCCAGGCGCGGGAACACTCAG3′ and 5′GGCGGACTTGCACTCGCTCTCG3′ for Arg213Gly in ECSOD. In the third primer pair one mismatch was induced in both primers: one to delete a secondary digestion site for MwoI and one to reduce the formation of secondary structures of the primer pair.
Polymerase chain reaction (PCR) amplifications were performed in 10–15 µl reaction volumes containing 20–50 ng genomic DNA, 2.5–6.0 pmol of each primer, 0.2 mM of each dNTP, and 0.2–0.4 U thermostable DNA polymerase (Dynazyme; Finnzymes, Espoo, Finland or AmpliTaqGold, Applied Biosystems, CA, USA) in the buffer recommended by the manufacturers with 1.5–2.5 mM MgCl2. The samples were denatured for 5–12 minutes at 94°C, subjected to 40–42 cycles each of 50 s at 94°C, 50 s at 58–64°C, and 1 minute at 72°C, and elongated for 8–10 minutes at 72°C. The reaction products were cleaved using restriction enzymes (New England BioLabs, Beverly, MA, USA), electrophoresed on 1.5–3% agarose gel, stained by Gelstar (FMC Bioproducts, Rockland, Maine, USA) and photographed under UV illumination.
The length of the PCR product for A>C substitution in the third intron of CuZnSOD was 278 bp for the major allele (A) and the length of the two HhaI digested fragments in the presence of the minor allele (C) were 71 and 207 bp. The length of the BsaWI digested PCR products for the Val16Ala allele of MnSOD was 249 bp and for the Val16 allele 87 and 162 bp. The length of the PCR product for Arg213Gly of ECSOD was 63 bp for the minor allele (Gly) and the lengths of the two MwoI digested fragments in the presence of the major allele (Arg) were 28 bp and 35 bp.
Genotyping was performed for Val16Ala in the whole data set and for A>C substitution and Arg213Gly in a random subset of samples (75 trios, N = 300 study individuals). The success rate in genotyping was 91.4% for Val16Ala, 99.6% for A>C, and 98.4% for Arg213Gly. Mendel errors were found in 0.4% of the genotypes. All the markers were in Hardy-Weinberg equilibrium.
Association analysis
For the association analysis, large pedigrees were divided into trios (mother-father-child) using an in-house computer program. The program identifies the maximal number of trios that are not overlapping and in which one or two members were affected (not both parents). Trios that included members who had not been genotyped or members with unknown phenotype were excluded. In case of ambiguities (missing genotype data, identical heterozygotic genotypes in all of the family members, or Mendel errors), the alleles were discarded. From each trio four independent chromosomes were obtained. If the child was affected, the transmitted alleles were considered as disease associated and the non-transmitted chromosomes as controls. If one of the parents was affected, his/her chromosomes were considered as disease associated and the spouse’s chromosomes as controls. If both the parent and the child were affected, only the non-transmitted chromosome of an unaffected parent was considered as the control and the other three as disease associated.
Statistical analysis
All allele comparisons were done using the χ2 test except when the expected number in a single cell in a 2 × 2 contingency table was less than five when the Fisher’s exact test was applied.
Power calculations
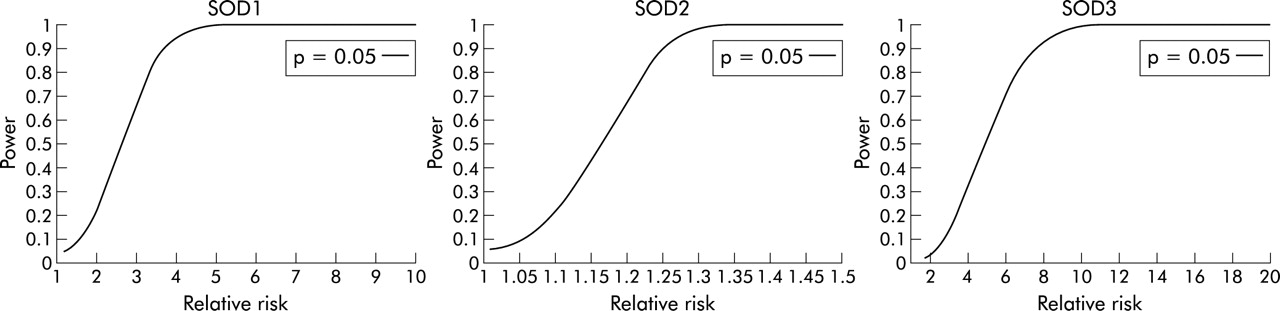
To assess the power of the sample size to detect an allele association, computer simulations were performed for each variant. In the simulations the observed prevalence of asthma in Finland, the size of the study cohort, and the frequencies of potential disease causing alleles estimated from the family based controls were fixed. One million iterations were performed. In each iteration the carrier status of all individuals was recomputed and a χ2 test was performed for the simulated frequencies. The power then corresponds to the proportion of iterations where the test yielded the selected significance threshold (p<0.05). As a consequence, the power (probability of observing the association between the variant and the disease status) was obtained for each SOD.
RESULTS
The allele distribution of SOD single nucleotide polymorphisms in asthma associated and family based control chromosomes retrieved from the trios is shown in table 1. None of the comparisons differed significantly from expected. The best nominal p value was computed for CuZnSOD (p = 0.09).
Allele distribution of SOD variants in asthma associated and family based control chromosomes
To assess the power of the sample set to detect an allele association, a computer simulation was performed by fixing the prevalence of asthma to 0.04 and by using the actual sample sizes studied for each gene (for MnSOD the whole data set and for CuZnSOD and ECSOD a random subset of samples). The frequencies of potential disease causing alleles in the population of the family based controls were estimated as follows: 0.100 for CuZnSOD (C at position 1934), 0.735 for MnSOD *Val16, and 0.034 for ECSOD *Gly213. By assuming these allele frequencies in the fixed sample size with the fixed prevalence of asthma, we can express the penetrance of carriers as a function of the penetrance of non-carriers or vice versa. Each combination of penetrances results in a different value of relative risk. Consequently, the power to detect the susceptibility allele can be expressed as a function of the relative risk. Based on the simulations, our study design had high power (>80%) to exclude a relative risk of more than 3.3 for CuZnSOD, more than 1.2 for MnSOD, and more than 6.6 for ECSOD at a p level of 0.05 (fig 1).
{kind=link}
Power to detect an association at a probability level of 0.05 between genetic variants in CuZnSOD, MnSOD, ECSOD and asthma as a function of relative risk. In the simulations the allele frequencies and prevalence of asthma in the population were fixed and actual sample sizes were used.
DISCUSSION
Experimental knockout models have shown that MnSOD in particular, but also ECSOD, play an essential role in protecting lung tissue against high oxygen tensions and exogenous oxidants.11 However, whether their common variants with functional implications have a significant role in the development of human lung diseases remains unknown. In our genome wide scan for asthma susceptibility loci performed in the same core pedigrees, no evidence for linkage was obtained at the chromosomal regions 6q25.3, 21q22.1, or 4p16.3–q21 where the SOD genes are located.29 Thus, none of these genes appears to be located in the neighbourhood with the most promising linkage peaks for asthma related traits. For the association analysis the data set was expanded with nuclear families. Allele frequencies for Ala16Val and Arg213Gly in the Finnish study population were comparable to those reported previously.15,23,24 Using this approach we were unable to find any evidence for an association between the three SOD variants and asthma.
These three variants show the complexity of the studies of common variants as potential disease causing alleles, since their frequency and therefore also the possible models of inheritance vary greatly. For common alleles such as the MnSOD variant, only extremely low penetrance models fit the inheritance (table 1). However, since they occur frequently in the population, they may have a significant role in the development of a disease even with a relative risk of less than 2. By studying this cohort, we could exclude (at 80% power and p = 0.05) all genetic models that would increase the risk among carriers by ⩾1.2 fold compared with the risk of asthma in the normal population. For the ECSOD and CuZnSOD variants the power to exclude genetic effects remained weaker (relative risk ⩾6.6 for ECSOD and ⩾3.3 for CuZnSOD). However, these variants are rare in the population (table 1) and therefore only the models of inheritance in which the penetrance (and thus also the relative risk) of an allele is high are of interest.
Based on our results, it is highly unlikely that these three variants of the SOD genes act alone as major players in the genetic susceptibility of asthma. With MnSOD and ECSOD we have focused on the variants which change the amino acid and which are functional. However, there is increasing evidence that genetic variation in the untranslated regions of the transcripts (effect on mRNA turnover) and intronic polymorphisms (effect on splicing of the gene) can be functionally important, both of which can be found in SOD loci. Our findings therefore do not exclude the possibility that, together with other impairments in the oxidant decomposing mechanisms (either genetic or environmental), SOD genes may become significant in inflammatory airway diseases where oxidant stress is evidently increased.
Acknowledgments
This study was supported by the Finnish Antituberculosis Association Foundation, Academy of Finland, and the Sigrid Juselius Foundation. Ms Satu Koljonen is acknowledged for her skilful technical assistance.