Article Text

Abstract
Background: Metallothionein (MT) is a protein that can be induced by inflammatory mediators and participate in cytoprotection. However, its role in inflammation remains to be established. A study was undertaken to determine whether intrinsic MT protects against acute inflammatory lung injury induced by bacterial endotoxin in MT-I/II knock out (−/−) and wild type (WT) mice.
Methods: MT (−/−) and WT mice were given vehicle or lipopolysaccharide (LPS, 125 μg/kg) intratracheally and the cellular profile of the bronchoalveolar lavage (BAL) fluid, pulmonary oedema, lung histology, expression of proinflammatory molecules, and nuclear localisation of nuclear factor-κB (NF-κB) in the lung were evaluated.
Results: MT (−/−) mice were more susceptible than WT mice to lung inflammation, especially to lung oedema induced by intratracheal challenge with LPS. After LPS challenge, MT deficiency enhanced vacuolar degeneration of pulmonary endothelial cells and type I alveolar epithelial cells and caused focal loss of the basement membrane. LPS treatment caused no significant differences in the enhanced expression of proinflammatory cytokines and chemokines nor in the activation of the NF-κB pathway in the lung between the two genotypes. Lipid peroxide levels in the lungs were significantly higher in LPS treated MT (−/−) mice than in LPS treated WT mice.
Conclusions: Endogenous MT protects against acute lung injury related to LPS. The effects are possibly mediated by the enhancement of pulmonary endothelial and epithelial integrity, not by the inhibition of the NF-κB pathway.
- IL, interleukin
- KC, keratinocyte chemoattractant
- LPS, lipopolysaccharide
- MCP-1, macrophage chemoattractant protein
- MIP-1α, macrophage inflammatory protein-1α
- MT, metallothionein
- NF-κB, nuclear factor-κB
- metallothionein
- lipopolysaccharide
- acute lung injury
- lipid peroxidation
Statistics from Altmetric.com
- IL, interleukin
- KC, keratinocyte chemoattractant
- LPS, lipopolysaccharide
- MCP-1, macrophage chemoattractant protein
- MIP-1α, macrophage inflammatory protein-1α
- MT, metallothionein
- NF-κB, nuclear factor-κB
Metallothionein (MT) is a highly conserved, low molecular weight, cysteine rich protein. Since cysteine residues of MT bind and store metal ions, it has been proposed that MT may have an important role in homeostasis and detoxication of heavy metals.1 It can react also with free radicals and electrophils because of its high sulfhydryl content,2,3 and can serve as a sacrificial scavenger for hydroxyl radicals in vitro4 and also protects against free radical induced DNA damage.5–7 In addition, MT is induced by heavy metals or oxidative stress producing chemicals,8 and exhibits cytoprotection against toxicity of heavy metals or alkylating anticancer drugs3 as well as against oxidative stress related organ damage.9,10 MT-I and MT-II genes are constitutively expressed in the liver and are highly induced by metals, glucocorticoids, and inflammatory mediators. Proinflammatory cytokines, including tumour necrosis factor (TNF)-α, interleukin (IL)-1, IL-6, and interferon-γ, induce hepatic MT gene expression in vivo. However, there are conflicting reports about the role of MT in inflammatory processes. In brief, MT plays a pivotal role in mediating the harmful effects of TNF induced shock11 but MT (−/−) mice have been reported to be more sensitive to lipopolysaccharide (LPS) induced lethal shock.12
Acute lung injury is characterised by neutrophilic inflammation, increased expression of proinflammatory cytokines, loss of epithelial and endothelial integrity, and the development of interstitial oedema.13–16 Intratracheal instillation of LPS produces a well recognised model of acute lung injury leading to the activation of alveolar macroghages, tissue infiltration of neutrophils, and interstitial oedema.17 Although inhalation of LPS has been reported to induce MT gene and protein in the lung in vivo,18,19 there is no evidence for the direct contribution of MT in acute lung injury elicited by LPS. The development of acute lung injury requires several pulmonary cell populations, transcriptional regulatory factors, and proinflammatory molecules. Nuclear factor-κB (NF-κB) activation and the subsequent expression of proinflammatory mediators also have an important role.20,21 However, the effects of MT on NF-κB activation is uncertain.22,23
This study was undertaken to determine whether intrinsic MT has a role in protecting against acute lung injury induced by LPS using MT-I/II null (MT (−/−)) mice and control wild type (WT) mice. The mechanisms by which MT protects against acute lung injury were also studied and its role in the NF-κB pathway in vivo determined.
METHODS
Animals and study protocol
MT (−/−) mice whose MT-I and MT-II genes had null mutation and WT mice were provided by Dr Choo (Murdoch Institute for Research into Birth Defects, Royal Children’s Hospital, Australia).24 They were of a mixed genetic background of 129 Ola and C57BL/6 strains. F1 hybrid mice were mated with C57BL/6 mice and their offspring were back-crossed to C57BL/6 for six generations in the National Institute for Environmental Studies (NIES; Tsukuba, Japan), reproducing normally and displaying no overt abnormality in physical state and behaviour. These mice were routinely bled in the vivarium of NIES as previously described.10
MT (−/−) and WT mice were treated with vehicle or LPS (E coli B55:05, Difco Lab, Detroit, MI, USA). For both genotypes the vehicle groups received 100 μl phosphate buffered saline (PBS) at pH 7.4 (Nissui Pharmaceutical Co, Tokyo, Japan) by intratracheal inoculation. The LPS groups received 125 μg/kg LPS dissolved in 100 μl of the same vehicle. Intratracheal inoculation was conducted as described previously.25,26
Lung water content, bronchoalveolar lavage (BAL), and measurement of cytokines and lipid peroxides
The lungs were weighed and dried as previously reported.27 The wet lung weight − dry lung weight/body weight was calculated (n = 5 in each group). BAL and cell counts were conducted as previously described (n = 5 in each group).26,28 Enzyme linked immunosorbent assays (ELISA) for IL-1β (Endogen, Cambridge, MA, USA), macrophage inflammatory protein (MIP)-1α (R&D Systems, Minneapolis, MN, USA), macrophage chemoattractant protein (MCP)-1 (R&D Systems), and keratinocyte chemoattractant (KC) (R&D Systems) in the lung tissue supernatants (n = 8 in each group) were carried out as reported elsewhere.26,28 Lipid peroxide in the lung was measured by a Determiner LPO kit (Kyowa Medics Co Ltd, Tokyo, Japan) (n = 8 in each group).
Microscopic examination
The lungs were fixed and stained with haematoxylin and eosin as previously described (n = 4 in each group).25 Lung tissue was fixed with 2.5% glutaraldehyde and rinsed in 0.1 M phosphate buffer (pH 7.4; n = 4 in each group). Ultrathin sections were stained with uranyl acetate and lead citrate and examined with a JEM-100CX electron microscope (JEOL, Tokyo, Japan).
Preparation of nuclear and cytoplasmic protein and Western blot analysis
Nuclear protein extracts (n = 5 in each group) were examined by Western blot analysis using a rabbit anti-p65 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the bands were quantitated as described previously.26
Statistical analysis
Data were reported as mean (SE) values except for the cellular profiles of BAL fluid and the lung water content using Stat View version 4.0 (Abacus Concepts Inc, Berkeley, CA, USA) as previously described.25 Differences in cellular profiles of BAL fluid, lung water content, and nuclear localisation of p65 subunit of NF-κB were analysed by a Kruskal-Wallis test followed by a Mann-Whitney U test using the SPSS 8.0 statistical package for Windows (Chicago, IL, USA). Differences in other data were analysed by ANOVA followed by Fisher’s PLSD test (Stat View version 4.0). p values of <0.05 were considered statistically significant.
RESULTS
Role of MT in acute lung injury induced by bacterial endotoxin
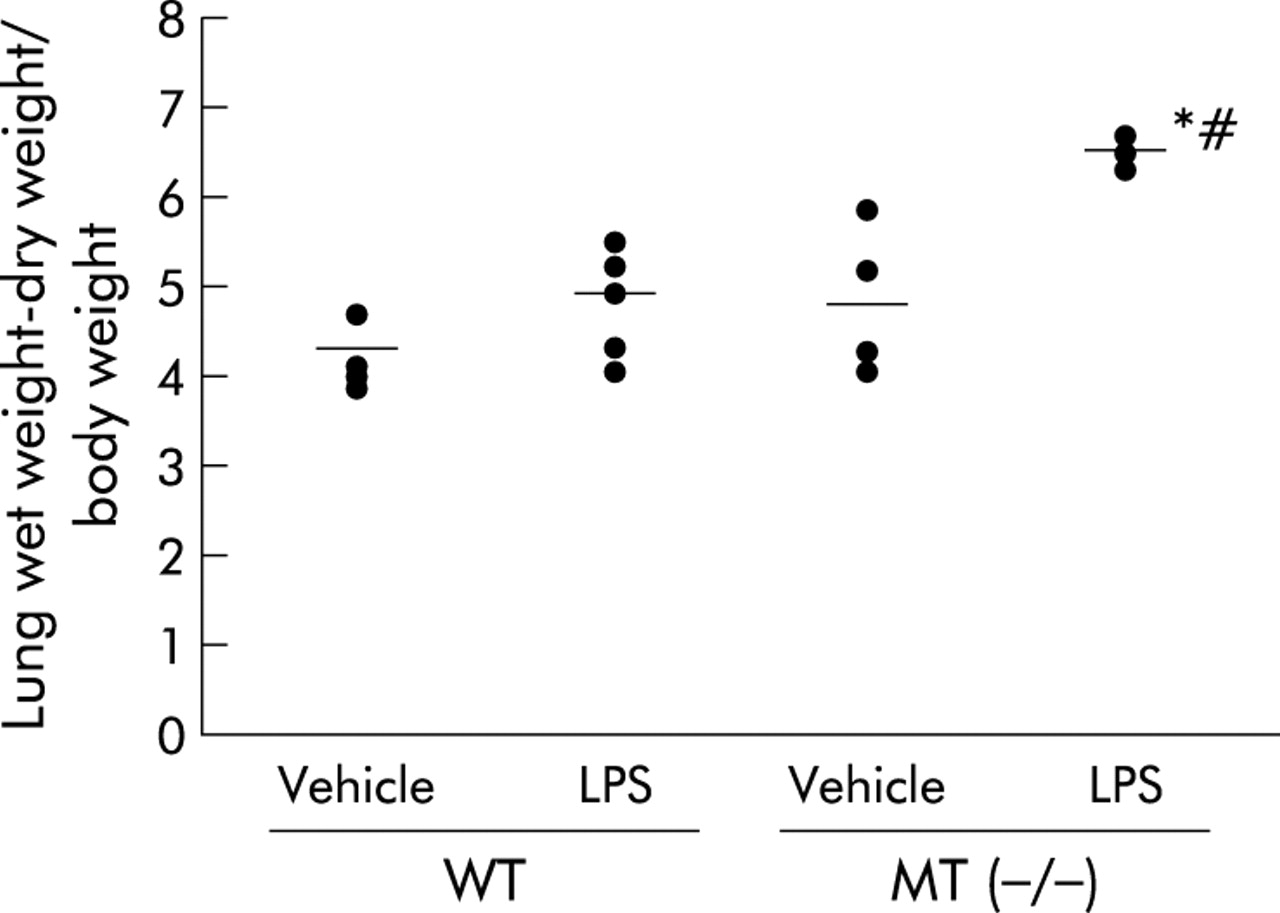
To examine the role of MT in pulmonary oedema related to bacterial endotoxin we measured the lung water content 24 hours after the intratracheal instillation of vehicle or LPS (fig 1). LPS increased the lung water content in both genotypes of mice compared with the vehicle (p<0.01 for MT (−/−) mice, not significant in WT mice). Following treatment with LPS, MT (−/−) mice had a significant and marked increase in the lung water content compared with WT mice (p<0.05).

Lung water content in wild type (WT) and metallothionein (MT)-I/II knock out (−/−) mice challenged with lipopolysaccharide (LPS, 125 μg/kg). The bilateral lungs were weighed immediately after exsanguination 24 hours following the intratracheal administration of LPS or vehicle, dried in an oven at 95°C for 48 hours, and the wet lung weight − the dry lung weight/body weight was calculated. The results are shown as mean (SE) values (n = 5). *p<0.01 v vehicle treated mice; #p<0.05 v LPS treated WT mice.
To determine the role of MT in neutrophilic lung inflammation induced by bacterial endotoxin, the cellular profile of BAL fluid 24 hours after intratracheal instillation was examined. In both genotypes of mice LPS treatment induced significant increases in the numbers of total cells (fig 2A) and neutrophils (fig 2B) compared with vehicle treatment (p<0.05). LPS treatment caused greater and significant increases in the number of total cells (fig 2A) and neutrophils (fig 2B) in the BAL fluid in MT (−/−) mice than in WT mice (p<0.05).

(A) Total cells and (B) neutrophils in bronchoalveolar lavage (BAL) fluid in WT and MT (−/−) mice challenged with LPS. Twenty four hours after the intratracheal administration of vehicle or LPS the lungs were lavaged for the analysis of BAL fluid. The total cell count was determined on a fresh fluid specimen using a haemocytometer. Differential cell counts were assessed on cytological preparations stained with Diff-Quik. The results are shown as mean (SE) values (n = 5). *p<0.05 v vehicle treated mice; #p<0.05 v LPS treated WT mice.
Effect of MT deficiency on LPS induced histopathological and ultrastructural changes in the lung
To determine the differences in the histological changes after LPS treatment in the presence or absence of MT, lung specimens stained with haematoxylin and eosin were examined 24 hours after intratracheal instillation. In the presence of LPS, WT mice showed a moderate infiltration of neutrophils (fig 3A) while in MT (−/−) mice there was a marked recruitment of neutrophils and interstitial oedema with alveolar haemorrhage (fig 3B, C). Vehicle administration alone caused no histological changes in either genotype (fig 3D, E).

LPS induced histopathological changes (A–E) and ultrastructural changes (F–I) in the lungs of WT and MT (−/−) mice. WT mice: (A) LPS, (D) vehicle; MT (−/−) mice: (B, C) LPS, (E) vehicle. The histopathological changes induced by LPS in WT mice showed moderate infiltration of neutrophils (A; arrows). LPS treatment showed more significant damage in the lungs of MT (−/−) mice which included recruitment of neutrophils (B; arrows) and alveolar haemorrhage (C). Original magnification ×320 (A, B, D, E), ×250 (C). LPS induced ultrastructural changes included the thickening of the alveolar basement membrane (arrow) in WT mice (I). LPS treated MT (−/−) mice had large vacuoles in the cytoplasm of endothelial cells and type I alveolar epithelial cells (F, G). Focal loss of the basement membrane (arrow) was seen in the alveolar walls of LPS challenged MT (−/−) mice (H). RBC, red blood cell; type I, type I alveolar epithelial cell; En, endothelial cell; Bm, basement membrane. Bar = 1 μm.
To elucidate the mechanisms by which MT protects against acute lung injury, especially against aggravated vascular permeability related to bacterial endotoxin, the ultrastructural changes in the lung were examined by electron microscopy 24 hours after intratracheal instillation. LPS treated MT (−/−) mice had large vacuoles in the cytoplasm of the endothelial cells and type I alveolar epithelial cells (fig 3F, G) accompanied by a focal loss of basement membrane in the alveolar walls (fig 3H). In LPS treated WT mice, on the other hand, only thickening of the alveolar basement membrane occurred (fig 3I). Following vehicle the alveolar wall was normal in both MT (−/−) and WT mice (data not shown).
Effect of MT deficiency on lung expression of proinflammatory cytokines and chemokines related to bacterial endotoxin
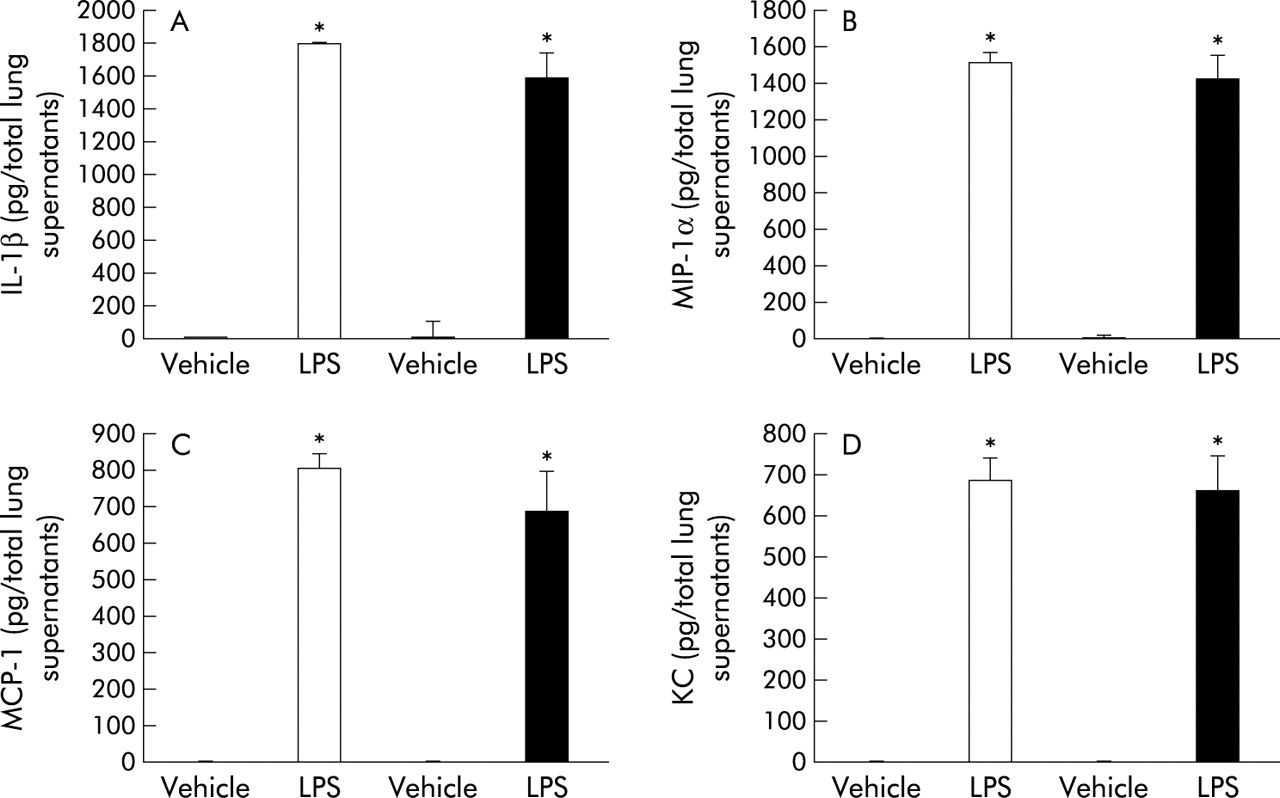
To investigate the role of MT in the lung expression of proinflammatory cytokines and chemokines related to bacterial endotoxin, we compared the protein levels of IL-1β, MIP-1α, MCP-1, and KC in lung tissue supernatants from the four experimental groups 24 hours after intratracheal instillation. LPS treatment induced significant increases in these cytokines and chemokines compared with vehicle treatment in both genotypes (fig 4A–D, p<0.01). In the presence of LPS treatment, however, the local expression of these cytokines and chemokines was not significantly different between the two genotypes (fig 4A–D).
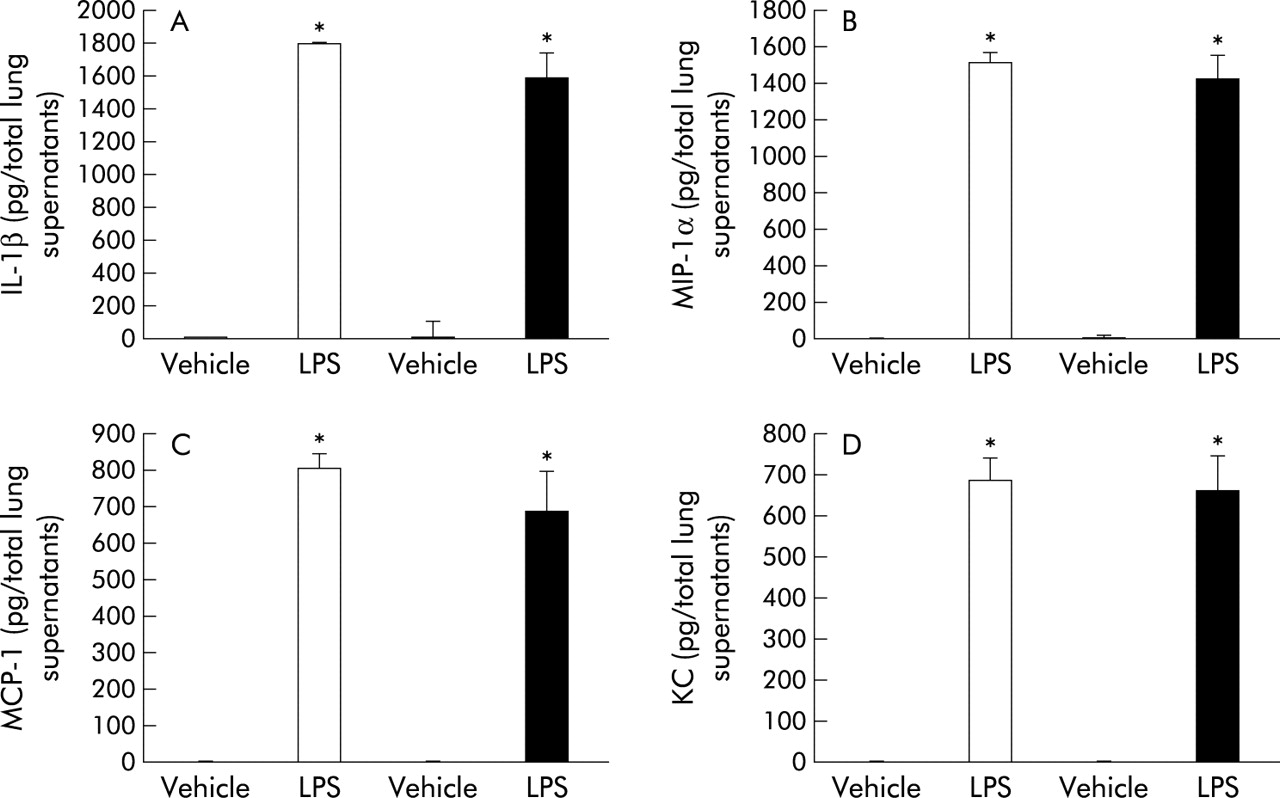
Protein levels of (A) interleukin (IL)-1β, (B) macrophage inflammatory protein (MIP)-1α, (C) macrophage chemoattractant protein (MCP)-1, and (D) keratinocyte chemoattractant (KC) in lung tissue supernatants 24 hours after challenge with vehicle or LPS (n = 8 in each group) measured by enzyme linked immunosorbent assays. White bar, WT mice; black bar, MT (−/−) mice. Results are mean (SE) values. *p<0.05 v vehicle treated mice.
Effect of MT deficiency on NF-κB activation related to bacterial endotoxin
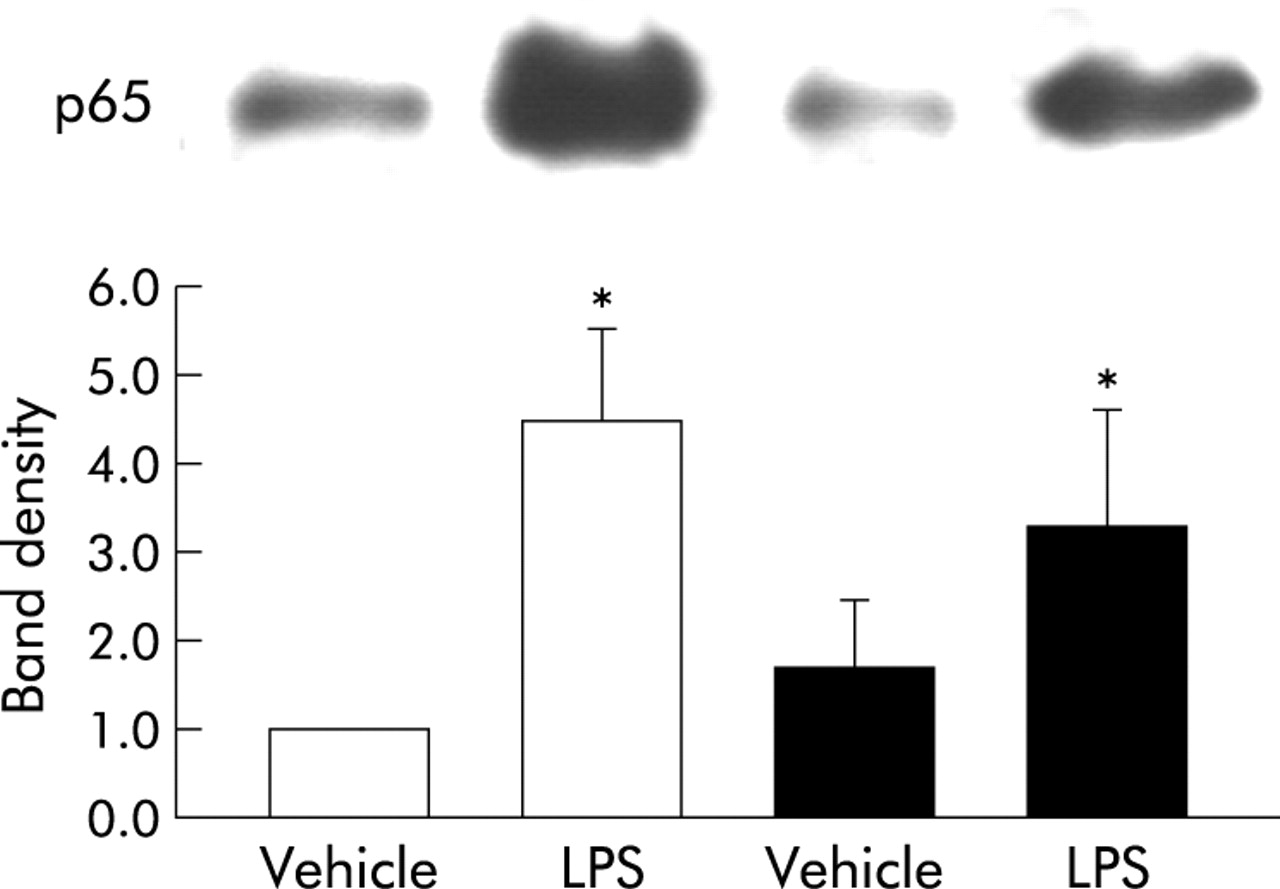
To determine whether the absence of MT affects the activation of NF-κB related to bacterial endotoxin in vivo, we compared nuclear protein levels of the p65 subunit of NF-κB in the lungs 2 hours following intratracheal instillation. In both genotypes LPS treatment significantly increased nuclear localisation of the p65 subunit of NF-κB compared with vehicle treatment (fig 5, p<0.05). However, there were no significant differences between the protein levels of MT (−/−) mice and WT mice after LPS administration (fig 5).

Nuclear localisation of the p65 subunit of nuclear factor-κB (NF-κB) in WT and MT (−/−) mice treated with vehicle or LPS. Nuclear localisation of the p65 subunit of NF-κB was investigated 2 hours after intratracheal administration using Western blot analysis. The top panel shows the actual membrane pictures of p65 and the bottom panel shows the band density for p65. White bar, WT mice; black bar, MT (−/−) mice. Each density represents the mean (SE) of at least five animals per group. *p<0.05 v vehicle treated mice.
Effects of MT on lipid peroxidation induced by bacterial endotoxin
We also examined the effects of MT on lipid peroxide formation in the lung. Compared with the vehicle, LPS increased the lipid peroxide content in both genotypes of mice (fig 6; p<0.05 for MT (−/−) mice, difference not significant in WT mice). LPS treatment led to a significantly greater increase in the lipid peroxide content in MT (−/−) mice than in WT mice (fig 6; p<0.05).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Formation of lipid peroxides in lung tissue supernatants determined by Determiner LPO kit. Values are mean (SE) of six animals per group. White bar, WT mice; black bar, MT (−/−) mice. *p<0.05 v other groups.
DISCUSSION
This study has shown that MT (−/−) mice are more sensitive to acute lung injury induced by intratracheal administration of LPS than WT mice, especially to pulmonary oedema. Electroscopic examination showed that LPS treatment causes vacuolar degeneration of pulmonary vascular endothelial cells and type I alveolar epithelial cells, and focal loss of the basement membrane in MT (−/−) mice but not in WT mice. The local expression of proinflammatory cytokines and chemokines and the activation of the NF-κB pathway in the lung induced by LPS were not significantly different in the presence or absence of MT. The lipid peroxide content in the lungs was significantly higher in LPS treated MT (−/−) mice than in LPS treated WT mice.
MT is a regulator of zinc and copper homeostasis. It has recently been reported that several inflammatory stimuli induce hepatic MT. The induction of MT is mediated by several cytokines such as IL-1, IL-6, TNF-α, and interferon-γ.19,29–33 More recently, it has been reported that MT (−/−) mice are protected from TNF induced lethal shock compared with WT mice.11 In addition, MT-I overexpressing mice are more sensitive to the lethal effects of TNF than WT mice.11 In contrast, Kimura and colleagues have reported that MT (−/−) mice are more sensitive to LPS induced lethal shock in GalN sensitised mice through the reduction of α1-acid glycoprotein.12 The specific role of MT in inflammation therefore remains controversial.
As for lung inflammation, inhaled LPS induces MT expression in vivo,18,19 which suggests that MT may play a role in lung inflammation induced by LPS. In particular, LPS induces MT in epithelial cells in the lung.18 We also confirmed that immunoreactive MT proteins in the lungs are found in the endothelial cells and alveolar epithelial cells of WT mice but not in those of MT (−/−) mice (data not shown). In this study we found that MT (−/−) mice were more susceptible to acute inflammatory lung injury induced by LPS than WT mice. In particular, pulmonary oedema induced by LPS was markedly enhanced by MT deficiency. Based on previous reports18,19 and the results of this study, it is suggested that MT may have protective properties against acute inflammatory lung injury induced by bacterial endotoxin, predominantly by protecting the integrity of the pulmonary vascular system.
Despite the sensitivity of the vascular endothelium to both heavy metal toxicity and oxidative stress,34,35 little is known about the role of MT in vascular endothelial/epithelial integrity. In human lungs immunoreactive MT is detected in pleural endothelial cells and basal cells from the bronchial epithelium.36 On the other hand, cultured sheep pulmonary artery endothelial cells which overexpress MT are resistant to pro-oxidant stimuli in a metal independent fashion.37 It has also been postulated that MT protects endothelial cells against oxidative stress with or without subsequent increased expression of cytokines, thrombin, and endothelin-1 in human venous endothelial cells.38
In the present study, electron histological examination of the lungs showed that LPS caused more pronounced damage of the vascular endothelial cells, endothelial basement membrane, and alveolar epithelial cells in MT (−/−) mice than in WT mice. It is likely that the protective role of MT in this model is mediated through the maintenance of endothelial and epithelial integrity. Since enhanced expressions of cyclo-oxygenase (COX)-239 and Rho kinase40 have been reported to play a role in endothelial retraction and permeability, we examined the local expression of these proteins by Western blotting (data not shown). In both genotypes LPS treatment increased COX-2 and Rho kinase proteins in the lung compared with vehicle treatment. However, there were no significant differences in the levels of these two proteins between MT (−/−) and WT mice after LPS administration (data not shown). Further studies are needed to elucidate the molecular mechanism by which MT protects the vascular integrity.
Activation of transcription factors such as NF-κB and the subsequent production of proinflammatory mediators play a critical role in the development of acute lung injury. NF-κB activation in the lung after intratracheal instillation of LPS is correlated with cytokine mRNA expression and neutrophilic alveolitis, supporting the idea that NF-κB activation is a pivotal event in the generation of neutrophilic lung inflammation.41 IL-1β, MIP-1α, MCP-1 and IL-8 have also been shown to participate in the pathogenesis of acute lung injury.42–45 Intriguingly, MT is observed in the nucleus and/or the cytoplasm of cells,46 suggesting that it can interact with nuclear transcription factors. In fact, TNF induced NF-κB activation in WT cells has been reported to be lower than that in MT (−/−) cells.23 Transfection of the MT-I gene to MT (−/−) cells reduces NF-κB activation by suppressing the degradation of I-κB, which suggests that MT functions as a negative regulator of NF-κB activation. In contrast, Abdel-Mageed and Agrawal22 have shown that MT potentiates the activation of NF-κB, so the apparent correlation between MT and NF-κB has not been established. In the present study MT deficiency did not alter the nuclear localisation of NF-κB. Although the levels of NF-κB induced proinflammatory molecules were increased in the lungs after LPS challenge, the differences between MT (−/−) and WT mice were not statistically significant. Our results indicate that the protective effect of MT on acute lung injury related to bacterial endotoxin may not be mediated via NF-κB dependent pathways. Further studies are needed to elucidate the role of MT in NF-κB mediated gene expression in other experimental models.
We have previously confirmed the expression of proinflammatory cytokines and chemokines including IL-1β, MIP-1α, MCP-1, and KC in the lung 24 hours after the intratracheal administration of LPS,26,28 and the localisation of NF-κB 2 hours after LPS administration.26 Close correlations were found between NF-κB activation, the enhanced local expression of proinflammatory mediators, and the magnitude of acute lung injury.26 We therefore examined the expression of proinflammatory mediators 24 hours after intratracheal LPS challenge and the activation of NF-κB 2 hours after LPS challenge in the present study. In addition, the overall trends for the expression of these proinflammatory proteins in the lung at other time points (2 and 6 hours after challenge) were similar to those at 24 hours after the intratracheal challenge in the present study (data not shown).
Oxidative stress is thought to play a role in the pathogenesis of acute lung injury.47 It has been reported that endotoxin treatment results in reduced glutathione levels in the lungs.48,49 Augmentation of the pulmonary antioxidant status can ameliorate LPS induced lung injury.50 MT can also assume the function of superoxide dismutase in yeast51 and protect against lipid peroxidation in erythrocyte ghosts produced by xanthine oxidase derived superoxide anion and hydrogen peroxide.52 One possibility is that the increased lung inflammation and pulmonary oedema in MT (−/−) mice result from the loss of antioxidative effects caused by MT deficiency. We therefore looked at the contribution of oxidative stress to the aggravation of LPS induced injury in the absence of MT. The immunoreactivity of 8-OhdG, pentosidine, N-(carboxymethyl) lysine (CML), and nitrotyrosine in the lungs of the four experimental groups were compared by immunohistochemistry (data not shown). 8-OhdG is a proper marker of oxidative DNA damage. Advanced glycation end products such as pentosidine and CML are reported to be the final products of oxidative stress.53–55 In addition, reactive nitrogen species have a number of inflammatory actions and their products such as nitrotyrosine are accurate biomarkers of oxidation of amino acids.56 In the presence of LPS challenge, however, the expression of these molecules did not differ between the two genotypes. Lipid peroxidation is another biomarker for oxidative stress. We have previously reported that lung injury caused by LPS is concomitant with the enhancement of lipid peroxidation in the lung.57 We therefore studied the effects of MT on lipid peroxide formation in the lung and found that the lipid peroxide content was higher in the lungs of LPS treated mice than in those of vehicle treated mice, both in the presence and absence of MT. In particular, LPS treatment resulted in significantly higher levels of lipid peroxides in the lungs of MT (−/−) mice than in those of WT mice. It is suggested that the enhancement of lipid peroxidation is involved, at least partly, in the aggravation of acute lung injury induced by LPS in MT (−/−) mice. Lipid peroxidation might also be related to the ultrastructural changes in the endothelial and epithelial cells seen in LPS treated MT (−/−) mice.
In conclusion, this study has shown that endogenous MT protects against acute lung injury induced by bacterial endotoxin possibly via a protective effect on the vascular integrity. The effect does not appear to be mediated through inhibition of the NF-κB pathway and subsequent proinflammatory cytokine expression. Augmentation of MT may provide alternative therapeutic strategies for acute lung injury, especially when treatments for inhibition of the NF-κB pathway are ineffective.