Article Text

Abstract
Background: Inhaled corticosteroids (ICS) affect many inflammatory pathways in asthma but have little impact on cysteinyl leukotrienes. This may partly explain persistent airway inflammation during chronic ICS treatment and failure to achieve adequate asthma control in some patients. This double blind, randomised, parallel group, non-inferiority, multicentre 16 week study compared the clinical benefits of adding montelukast to budesonide with doubling the budesonide dose in adults with asthma.
Methods: After a 1 month single blind run in period, patients inadequately controlled on inhaled budesonide (800 μg/day) were randomised to receive montelukast 10 mg + inhaled budesonide 800 μg/day (n=448) or budesonide 1600 μg/day (n=441) for 12 weeks.
Results: Both groups showed progressive improvement in several measures of asthma control compared with baseline. Mean morning peak expiratory flow (AM PEF) improved similarly in the last 10 weeks of treatment compared with baseline in both the montelukast + budesonide group and in the double dose budesonide group (33.5 v 30.1 l/min). During days 1–3 after start of treatment, the change in AM PEF from baseline was significantly greater in the montelukast + budesonide group than in the double dose budesonide group (20.1 v 9.6 l/min, p<0.001), indicating faster onset of action in the montelukast group. Both groups showed similar improvements with respect to “as needed” β agonist use, mean daytime symptom score, nocturnal awakenings, exacerbations, asthma free days, peripheral eosinophil counts, and asthma specific quality of life. Both montelukast + budesonide and double dose budesonide were generally well tolerated.
Conclusion: The addition of montelukast to inhaled budesonide is an effective and well tolerated alternative to doubling the dose of inhaled budesonide in adult asthma patients experiencing symptoms and inadequate control on budesonide alone.
- asthma
- add-on therapy
- leukotriene receptor antagonists
- montelukast
- budesonide
Statistics from Altmetric.com
Chronic inflammation is recognised as a central component of asthma pathophysiology.1,2 Invading inflammatory cells in lung tissue release a wide variety of mediators and cytokines that contribute to the clinical characteristics of asthma.3 Cysteinyl leukotrienes released from eosinophils and mast cells are important pro-inflammatory asthma mediators which give rise to bronchoconstriction, mucus secretion, increased vascular permeability, smooth muscle hypertrophy, and inflammatory cell infiltration.4
Inhaled corticosteroids (ICS) affect a variety of inflammatory pathways in asthma and represent a gold standard in anti-inflammatory treatment.2 However, for some patients with persistent asthma, ICS as prescribed may fail to achieve adequate control. Increasing the ICS dose is one therapeutic option but clinical trials suggest this option may only help a proportion of patients3 and concerns exist that high dose ICS may be associated with local and systemic side effects.4,5 These issues have led to trials of adding other agents to ICS rather than increasing the dose, with results suggesting in the patient groups studied that adding other agents such as inhaled long acting β agonists may achieve at least similar benefits to increasing inhaled steroids.3,6,7 The principle that increasing inhaled steroids is only one option in patients with uncontrolled asthma receiving ICS alone has become accepted in guidelines.2
Research into the pathogenesis of asthma has led to the development of specific anti-inflammatory treatments, including montelukast, which blocks the interaction of cysteinyl leukotrienes with their receptor and resulting downstream events. Since montelukast attenuates leukotriene mediated effects, combination therapy with montelukast and ICS represents a theoretical alternative to increasing the ICS dose in patients inadequately controlled on ICS alone. Although several studies have demonstrated additive effects of montelukast with ICS,8–10 none have compared this effect with higher dose ICS as has been done with other treatments.3 A double blind non-inferiority randomised 16 week study was therefore performed to compare the clinical benefits of adding montelukast to inhaled budesonide with doubling the dose of inhaled budesonide in adult patients who were symptomatic on inhaled budesonide alone.
METHODS
Study population
Patients enrolled in the study were non-smokers or ex-smokers (stopped for at least 6 months and <12 pack year history) diagnosed with asthma for >1 year, aged 15–75 years, who were not optimally controlled as judged by investigators in spite of a regular ICS prescription at doses of 600–1200 μg/day for budesonide, beclomethasone, triamcinolone, flunisolide, and 300–800 μg/day for fluticasone. Patients were required to have forced expiratory volume in 1 second (FEV1) values ≥50% predicted at visits 1 and 3, together with ≥12% improvement in FEV1 after β agonist administration, and symptoms requiring β agonist treatment of at least 1 puff/day during the last 2 weeks of the run in period.
Patients were excluded if they had other active pulmonary disorders, respiratory infection within 3 weeks of visit 1 or during the run in period, treatment in an emergency setting within 2 months of visit 1, systemic corticosteroid treatment within 1 month, cromones or leukotriene receptor antagonists within 2 weeks, long acting antihistamine within 1 week (astemizole 3 months), or long acting β agonists or anticholinergic agents within 24 hours. The study was approved by the appropriate ethical review committees and each patient gave written informed consent.
Study protocol
The study included a 4 week run in period during which patients were switched to budesonide Turbohaler (800 μg/day (200 μg, two puffs twice daily). After 1 week single blind montelukast placebo was added; β agonist use and daytime symptoms were assessed during this period to determine eligibility for randomisation and to establish baseline values. Patients were randomised to one of two treatment groups for 12 weeks. Group 1 (MONT-BUD) received montelukast 10 mg/day (one tablet at bedtime) in addition to budesonide 800 μg/day and group 2 (BUD1600) received budesonide 1600 μg/day (800 μg twice daily) while receiving oral placebo montelukast. Budesonide in both groups was identical in appearance. Patients were instructed to withhold inhaled β agonist (for 6 hours) and short acting antihistamines (within 48 hours) before clinic visits (every 4 weeks).
Morning peak expiratory flow (AM PEF) was the prespecified primary end point. Other prespecified end points included initial treatment effect (days 1–3 AM PEF) and time course of morning PEF during the first 14 days of double blind treatment, daily self-reported β agonist use, daytime symptoms, nocturnal awakenings, asthma exacerbations, asthma free days (defined as any day free of oral corticosteroid use, emergency care, nocturnal awakenings, with use of ≤2 puffs of β agonist), peripheral blood eosinophil counts, asthma specific quality of life, and resource utilisation. Patients assessed daytime asthma symptoms in the evening before bedtime using a validated diary card containing four questions (scored from 0 to 6 where 0 is best).11 Patients also recorded nocturnal awakenings and overnight β agonist use.
A day with an asthma exacerbation was defined as a day with: a decrease from baseline in AM PEF of >20% or AM PEF <180 l/min or an increase in β agonist use of >70% (minimum increase of two puffs), or an increase in symptom score of ≥50%, or an asthma attack (worsening of asthma requiring an unscheduled visit to the doctor’s office, emergency room, admission to hospital, or treatment with oral corticosteroids). Patients were allowed to use short acting β agonists on an “as needed” basis but were encouraged to use only the amount required. At baseline and week 12 (or on withdrawal from the study) patients completed a validated self-administered asthma specific quality of life questionnaire (questions scored from 1 to 7 where 1 is worst).12
Safety
Adverse experiences were monitored throughout the study. Investigators evaluated all clinical adverse events in terms of intensity, duration, seriousness, outcome, and relation to test drugs.
Statistical analysis
The efficacy analyses were performed using an intention-to-treat approach, including all randomised patients with at least one baseline measurements and one after randomisation. The primary end point of change from baseline in AM PEF was assessed by an analysis of variance (ANOVA) model with terms for treatment and study centre.
The primary hypothesis of non-inferiority was tested by constructing the 95% two sided confidence interval (95% CI) around the treatment differences derived from the model. Montelukast added to budesonide was considered as effective as a double dose of budesonide if the two sided 95% confidence interval of the treatment difference (change from baseline on budesonide double dose minus change from baseline on montelukast + budesonide) was entirely below 10 l/min. Change from baseline in AM PEF was calculated as the difference between the mean of all AM PEF measurements obtained during the last 10 weeks of the study (that is, excluding the first 2 weeks after randomisation) and the baseline mean PEF value (since the higher dose ICS treatment effect may take 1–2 weeks to manifest itself). A sample size of 676 patients was calculated to provide 90% power to demonstrate non-inferiority.
The time course of the treatment effect during the first 14 days of double blind treatment was studied graphically. Other continuous end points such as initial change (first 3 days after starting study medication) in AM PEF, “as needed” β agonist use and symptom score, and overall change (during the entire 12 week treatment period) in quality of life, peripheral eosinophil count, “as needed” β agonist use, and daytime symptom score were analysed using a similar ANOVA model as for the primary end point. For percentage of days with asthma exacerbations, asthma free days, and nocturnal awakenings the analysis was performed after transformation to normal scores. The time course in AM PEF values was further explored by post hoc analysis using mixed model methodology13 with random effects for both intercept and time. The covariance was modelled using the autoregressive structure. The time course during the run in period (last 2 weeks before randomisation) was modelled with fixed effects for centre and time. The time course during the last 10 weeks of the double blind treatment period was modelled using fixed effects for centre, treatment, and time per treatment. A similar model was used over this time period (weeks 2–12), adding an extra variable to investigate a potential plateau in AM PEF at week 10. Binary end points such as resource utilisation and incidence of adverse events were compared using Fisher’s exact test.
RESULTS
Patients
A total of 1192 subjects were screened and 889 patients randomised, 448 to the MONT-BUD group and 441 to the BUD1600 group; 46 patients withdrew after randomisation (20 MONT-BUD, 26 BUD1600) because of clinical adverse events (n=20), protocol deviations (n=10), lost to follow up (n=8), withdrew consent (n=6), or personal reasons (n=2). There were no clinically meaningful differences between the groups in baseline characteristics (table 1), incidence of concomitant diseases, or use of concomitant drug treatments (data not shown).
Baseline characteristics of randomised patients
Efficacy
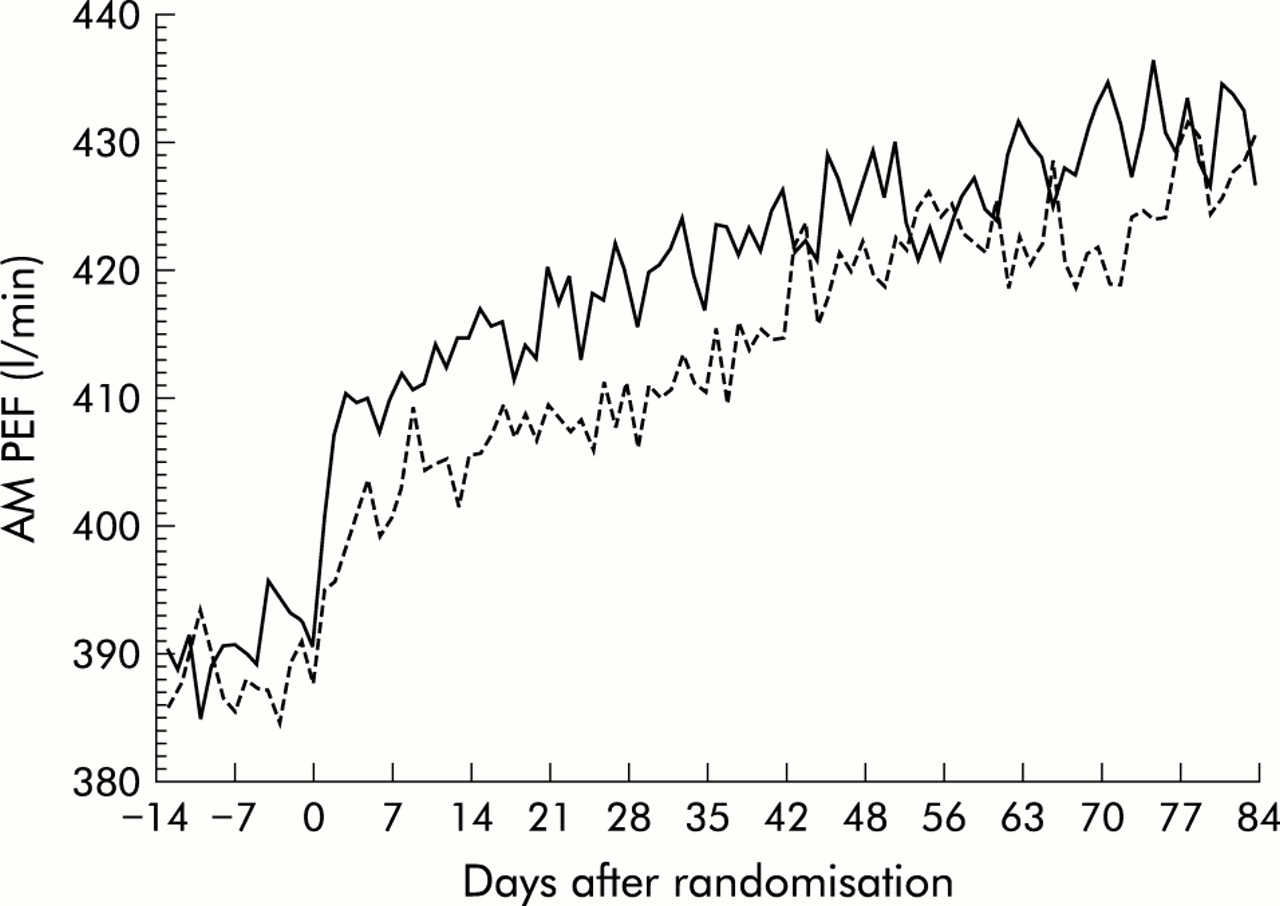
Both treatment groups had significant and progressive improvements in AM PEF compared with baseline over 12 weeks (fig 1). The improvement in AM PEF over the last 10 weeks of the 12 week treatment period with the addition of montelukast was at least as effective as doubling the budesonide dose (33.5 l/min v 30.1 l/min in the MONT-BUD and NUD1600 groups, respectively; 95% CI –12.9 to 4.8 for the difference). The distribution of response which was similar for both treatment arms is shown in fig 2. These findings were consistent across a variety of subgroups including sex, age, race, prior corticosteroid dose, baseline FEV1, baseline β agonist use, and concomitant allergic rhinitis or nasal polyps. The initial change from baseline in AM PEF during the first 3 days of treatment was rapid in onset and significantly greater in the MONT-BUD group than in the BUD1600 group (20.1 l/min v 9.6 l/min; 95% CI –17.6 to –4.3, p<0.001, fig 3). Beta agonist use (–0.63 v –0.44 puffs/day, 95% CI for difference 0.03 to 0.44, p<0.05) and daytime symptom improvement (–0.21 v –0.14, 95% CI for difference –0.02 to 0.19, p>0.05) followed the same pattern.
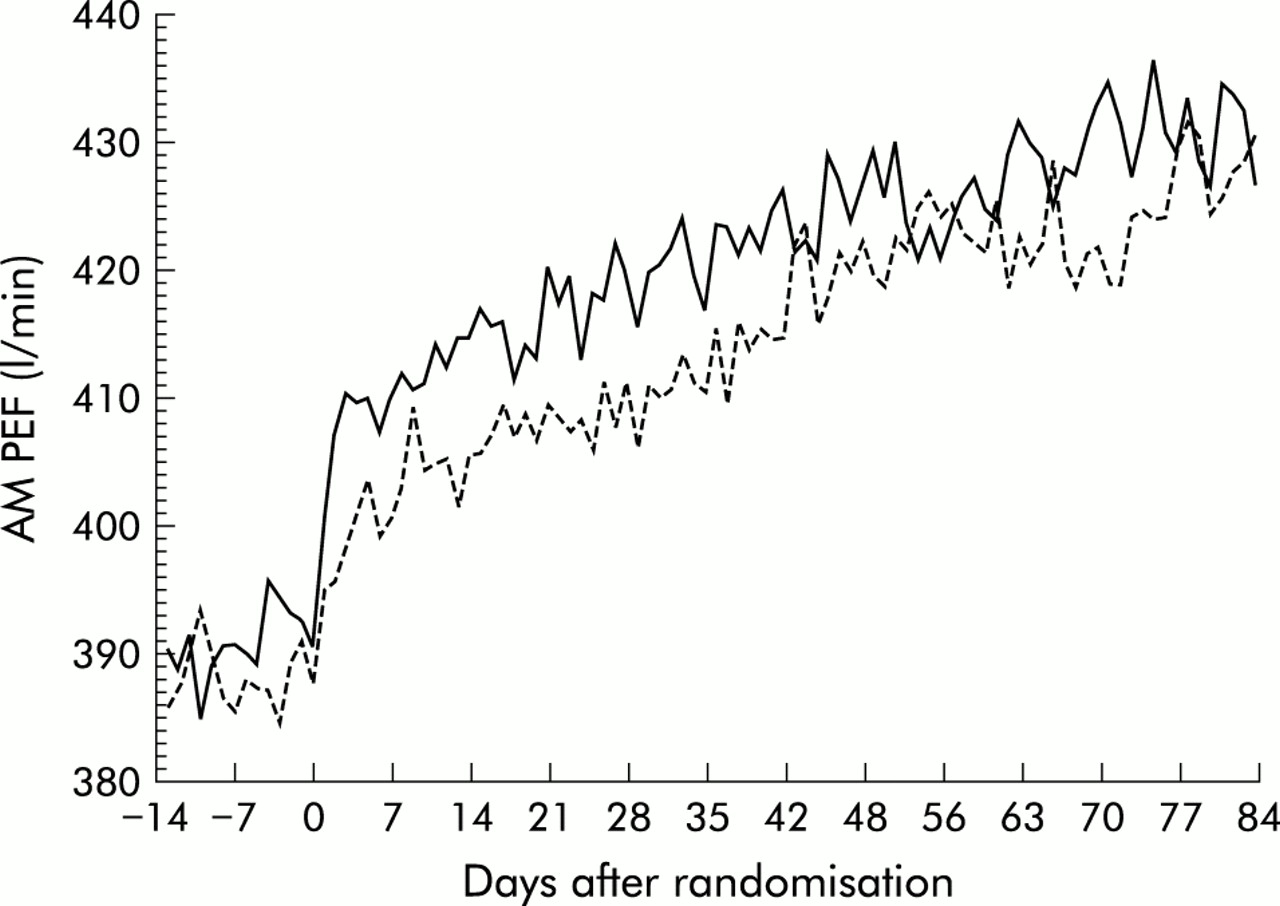
Morning peak expiratory flow (AM PEF) over the 12 week treatment period (solid line=montelukast + budesonide 800 μg daily, dashed line=budesonide 1600 μg daily). Data represent the mean AM PEF measured before administration of study medication.
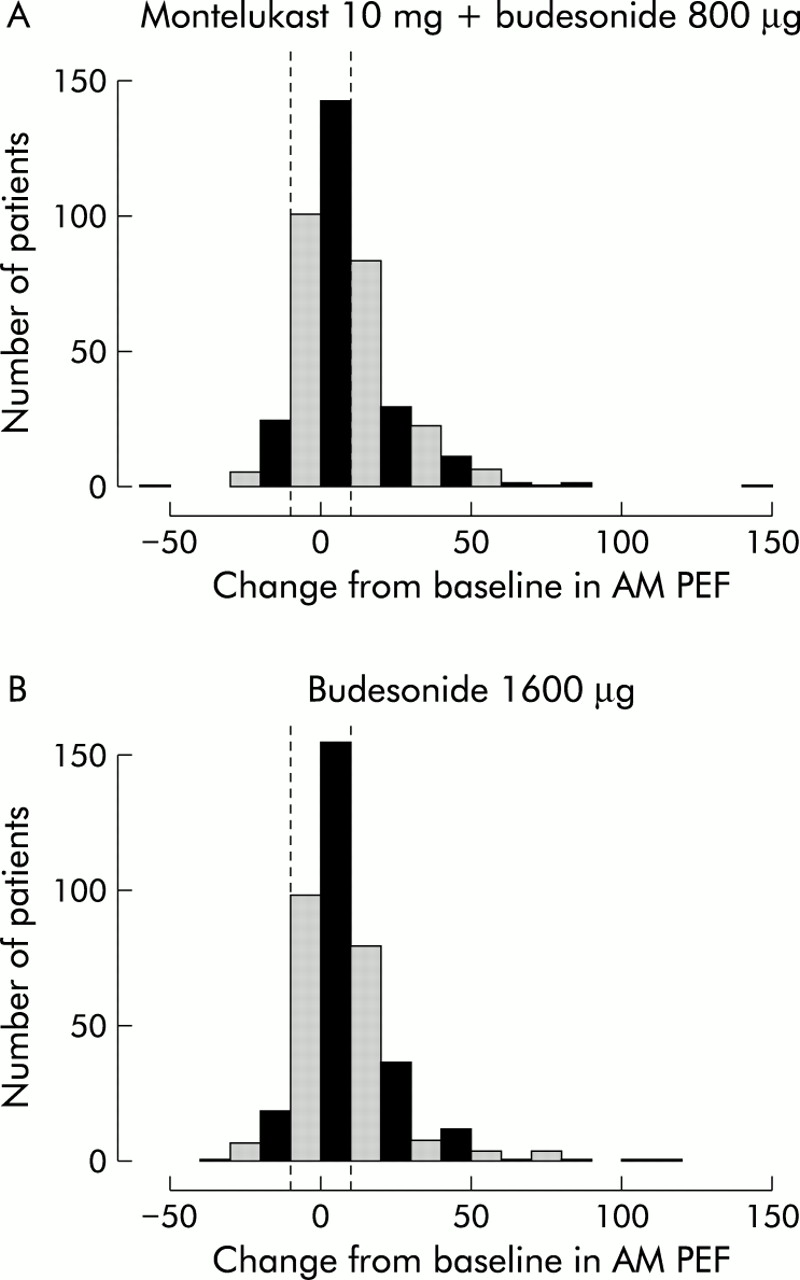
Distribution of response in mean change from baseline in morning peak expiratory flow (AM PEF) in (A) patients treated with montelukast + budesonide 800 μg daily and (B) those treated with budesonide alone in a dose of 1600 μg/day.

(A) Morning peak expiratory flow (AM PEF), (B) daily β agonist use, and (C) daytime symptom score measured daily during the last 14 days of the single blind placebo run in period and during the first 14 days of active treatment with either montelukast + budesonide (800 μg daily, solid line) or budesonide alone (1600 μg/day, dashed line). Data represent the mean values on each day.
AM PEF increased progressively during the last 10 weeks of the double blind treatment period in the MONT-BUD group (1.88 l/min per week, p<0.001) and in the BUD1600 group (2.41 l/min per week, p<0.001) but remained unchanged during the run in period (p=0.096). There was no evidence that the increase in AM PEF levelled off by the end of the study in either treatment group (p>0.05).
Progressive improvements over the 12 weeks after randomisation compared with no improvement during the run in period were evident in terms of as needed β agonist use and daytime symptom score (fig 4). Changes from baseline in β agonist use were –0.78 and –0.75 puffs per day for the MONT-BUD and BUD1600 groups, respectively (p=0.510), and in daytime symptom score were –0.34 and –0.35 in the two groups (p=0.908). Patients in both treatment groups improved and were not significantly different with respect to improved nocturnal awakenings with asthma (from 12.3% to 2.3% and from 13.8% to 3.9% of nights in the MONT-BUD and BUD1600 groups, respectively, p=0.353), median days with asthma exacerbations (6.7% v 6.3%, p=0.781), proportion of patients requiring oral steroids or admission to hospital (1.6% v 2.3%, p=0.472), median asthma free days (86.7% v 82.2%, p=0.371), change from baseline in peripheral blood eosinophil count (–0.05 v –0.07 × 109, p=0.387), and number of withdrawals because of asthma (2 v 7, p=0.105). Median days with asthma exacerbations fell when the first 4 weeks after randomisation were compared with the subsequent two 4 week periods in both groups (6.1 and 7.4 days in the first 4 weeks; 3.6 and 3.6 in the second 4 weeks; 3.2 and 3.3 in the final 4 weeks).

(A) Change in daily β agonist use and (B) daytime symptom score over the 12 week treatment period (solid line=montelukast + budesonide 800 μg daily, dashed line=budesonide 1600 μg daily). Data represent mean (SE).
Patients in the MONT-BUD and BUD1600 groups experienced similar significant improvements from baseline in each of the four domains of the quality of life questionnaire (fig 5). The change from baseline in overall asthma specific quality of life averaged 0.71 for MONT-BUD patients and 0.59 for BUD1600 patients, a between group difference approaching statistical significance (p=0.091). Self-reported treatment adherence was high in both groups for both tablets and inhalers with >95% of days reported as fully compliant with treatment. As with the other end points, there was a distribution of response in quality of life.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Mean change in quality of life (QOL) score including overall score and individual domains of patients treated with montelukast 10 mg/day + budesonide 800 μg/day (shaded bars) or budesonide 1600 μg/day (solid bars). (B) Net percentage of patients with clinically important changes in quality of life (calculated as proportion of patients improving by ≥0.5 points minus patients worsening by ≥0.5 points).
Safety
Both treatment regimens were generally well tolerated with no significant differences in the number of patients with adverse events, drug related adverse events, serious adverse events, or discontinuing treatment because of adverse events. There were significantly fewer investigator diagnosed respiratory adverse events in the MONT-BUD group than in the BUD1600 group (11.6% v 16.6% of patients, p<0.05). In the MONT-BUD group 166 patients (37.1%) experienced an adverse event compared with 182 patients (41.3%) in the BUD1600 group. The most common adverse events were upper respiratory infection, asthma worsening, and headache.
DISCUSSION
This double blind randomised 16 week study in asthma patients symptomatic while receiving budesonide 800 μg/day has shown that the addition of montelukast to budesonide produced comparable and substantial improvements in asthma control compared with doubling the dose of budesonide. However, during the first 3 days of treatment montelukast + budesonide was associated with a faster onset of action, as evidenced by a significantly greater change in AM PEF and reduction in β agonist use. In addition, both groups showed comparable and progressive improvements in other end points including as needed β agonist use, daytime symptom score, nocturnal awakenings, asthma exacerbations, asthma free days, blood eosinophil counts, and asthma specific quality of life.
An interesting finding of this study was the progressive steady increase in AM PEF during the course of the 12 week treatment period in both study groups. Indeed, at the end of 12 weeks of treatment a clear plateau had not been reached. This is surprising for patients in whom montelukast had been added to the treatment regime, although these findings are consistent with a recent 6 week study by Virchow and colleagues who also found that AM PEF continued to rise on a week by week basis following treatment of asthma patients with high dose zafirlukast and high dose ICS, while no such improvement occurred in the placebo arm.14 This does, however, differ from a previous study of montelukast + ICS where an early plateau was reached.8 One explanation for this difference is that our study population might have had more severe asthma with persisting symptoms and impaired lung function in spite of a higher prestudy ICS dose, much as in the study by Virchow. This suggests a mechanism of action that is not seen in patients with milder asthma, possibly similar to that seen with higher dose inhaled steroids. The clinical implication of these results, taken with the data from Virchow et al, is that in patients with more difficult asthma we need to consider longer trials of treatment of at least 12 weeks if using a leukotriene antagonist.
The results in the group who received an increased dose of budesonide are equally intriguing, suggesting that maximal benefit is not achieved even by 12 weeks. They also appear to run counter to the idea of a very flat dose-response curve for inhaled steroids.15 It is interesting that additional benefits of high dose ICS have also been observed in several other studies.6,7,16,17 The reason for this discrepancy may be similar to that for the montelukast group. These were symptomatic patients with asthma which was significantly reversible in spite of being prescribed inhaled steroids at approximately 800 μg/day—that is, they had room to improve. The activity of high dose ICS may also be different in that it may treat certain aspects of disease such as airways hyperresponsiveness and airway wall remodelling more effectively.17,18 Many of the dose ranging studies may fail to show this effect because they include patients who are completely controlled with low dose ICS and thus may not be powered to show a response above such doses. Meta-analyses may also not show such effects as they may lack high dose studies in appropriate patients.15
The reductions in the number of days with exacerbations were progressive and similar in the two groups, consistent with the effects of montelukast and higher dose ICS in other studies.11,19 The steady increase in pulmonary function and the progressive decrease in exacerbations observed in our study cannot be explained by simple bronchodilation and may be consistent with the reported anti-inflammatory effects of montelukast in several indices of asthmatic inflammation, including eosinophil counts in peripheral blood, sputum, and lung tissue20–24 and decreased exhaled nitric oxide.25 Based on these findings, additional studies involving measurement of inflammatory markers such as sputum eosinophils and longer study durations will be needed to explore this phenomenon further.
In addition to its effect on pulmonary function, asthma affects the physical, social, and emotional aspects of patients’ lives. In this study considerable improvement was seen in all domains of an asthma specific quality of life questionnaire (symptoms, activity, emotional function, and environmental stimuli) which were similar in both treatment groups, ranging from 0.56 to 0.71.12 These mean changes are greater than the individual difference of 0.5 prespecified for this study (and reported elsewhere) as the minimally clinically important difference for the asthma related quality of life questionnaire.26 It is also notable that a marked variation in response was seen between individuals for quality of life and other outcomes which are consistent with results in other studies.3,8
Both treatment approaches in this study achieved clinically significant control of asthma as evidenced by a low percentage of exacerbation days (median <7%; high rates of asthma free day (median >82%); progressive reduction in rescue β agonist use and symptoms; infrequent respiratory adverse events (lower in the montelukast group); and important improvements in nocturnal waking and quality of life. Both montelukast + budesonide and double dose budesonide were generally well tolerated. The incidence and type of adverse reactions reported for montelukast were consistent with the generally favourable tolerability profile of this drug in children and adults.27 The study was neither designed nor powered to observe differences in infrequent adverse events including those related to higher doses of ICS.
One potential limitation of the present study is the absence of a placebo group. However, during the last 2 weeks of the placebo run in period all patients received inhaled budesonide and a daily montelukast placebo tablet in a single blind fashion. Subsequently, on day 1 of the treatment phase patients were unaware that the montelukast placebo had been switched to active drug or that the ICS dose was doubled. The flat baseline, faster onset of action in the MONT-BUD group, and abrupt increase in AM PEF observed in both treatment groups is consistent with a true therapeutic effect rather than a placebo effect or regression to the mean, which would have been expected to manifest itself as a gradual rise in AM PEF beginning during the placebo run in period and continuing throughout both treatment periods of the study. In view of the flat baseline and persistent symptoms, it would also have been difficult to justify ethically maintaining patients on treatment that was not improving their asthma.
Patients with persistent asthma symptoms are typically managed by increasing the dose of ICS or adding a second therapeutic agent. Increasing the dose of ICS may be associated with a number of potential side effects4,5 and higher doses may not necessarily result in more effective control of asthma symptoms for all patients.3,28,29 International guidelines therefore recommend that the ICS dose should be minimised whenever possible.2 The addition of a second controller agent with a complementary mechanism of action may therefore be appropriate. The findings of the present study suggest that the addition of montelukast to ICS offers comparable asthma control to doubling the dose of ICS with a faster onset of action, and might lessen the potential risk of side effects associated with long term administration of high dose ICS.
APPENDIX
The members of the montelukast COMPACT study group are as follows: Argentina: R Alchapar, C E Baena-Cagnani, A M Lopez, L J Nannini, H Neffen, O Callejas, C A Rey, R Rojas, J Taborda; Australia: D Bryant, C Dennis, P Holmes; Austria: J Eckmayr, F Kummer, H Obermair, M G Roger, N Vetter, W Wanka, M Wild, C Wildner, H Zwick; Belgium: H Dierickx, J Mestdagh, L Rombouts, E Schatteman, H Schoofs; Brazil: J Fiterman, S M Barreto; Canada: M Laviolette; China: J Lin, W Luo, N S Zhong, X Zhou; Colombia: F Naranjo; Finland: O Rossi, T Toljamo, M Torkko; Germany: J Becker, K M Beeh, U Harnest, A Linnhoff, P Mikloweit, W Mitlehner, S Schmidtmann, R Schultebraucks, C Seevers, P Stutz; Greece: P Christaki, A Gaga, N Galanis, K Gourgoulianis, C Gratsiou, S Konstantopoulos, N E Papadakis, G Papageorgiou, N Siafakas; Hungary: Z Baliko, G Berta, Z Csontos, I Herjavecz, P Kraszko, K Nekam, M Namenyi, P Magyar, Z Szucs; Italy: S Centanni, G U Di Maria, L M Fabbri, G Schmid; Mexico: M Acuna, D Hernandez, R Perez, R Salazar; Netherlands: R Aalbers, T Bantje, A J M Bax, J P H M Creemers, A Harreveld, K van Kralingen, A Kuipers, P Luursema, J van Noord, L H M Rijssenbeek-Nouwens, H E J Sinninghe Damste, B M van Weelden, A M J Wever; Norway: A Bergmann, B Johansen, K E Langaker; Portugal: J Almeida, A M Arrobas, M S Miranda, A L Segorbe; Romania: J R Radu, I P Stoicescu; Spain: I A Ercoreca, J C Hernandez, J M Olaguibel-Rivera, A Pelaez, J L Prieto-Andres, F G Reques, J R Rodriguez, C C Sanz; Sweden: G Johansson, A Lind, B Lundback, A Spjuth; Switzerland: H J Graf, P Gumowski, M Pons, F Spertini; Taiwan: C D Chiang, J H Wang; UK: N Barnes, P Corris, B O’Connor, P Dilworth, M Fahmy, P Fletcher, I G James, D B Price; Venezuela: M D C Montes De Oca.
Acknowledgments
This study was supported by a grant from Merck & Co Inc, Whitehouse Station, New Jersey, USA. The authors thank Veerle Coenen for coordination of study data, Pam S Dellea for study coordination, and Dr Brigitte Frischer for protocol development, Samantha Knowles and Margaret Ross for secretarial support and Drs P Polos, D Yin, M Thomas, C Hand, and L Shepstone for their helpful comments. They also acknowledge the excellent study monitors, investigators, and patients.
REFERENCES
Footnotes
-
David Price and Jon Leff undertook the initial study design, protocol production, co-led the evaluation, co-wrote the data analysis plan, reviewed the statistics report and prepared the manuscript for publication. Leen Gilles contributed to the study design, co-wrote the data analysis plan, undertook the statistical analysis and contributed to the manuscript. All other authors reviewed and contributed to study design, protocol review, patient recruitment, data analysis review and input into the manuscript. David Price, Leen Gilles and Jon Leff are guarantors for the paper.
-
Conflicts of interest: David Price either through his role at the University of Aberdeen or personally has received grants, honoraria or educational support from the UK NHS R&D programme, 3M Pharmaceuticals, Abbot Laboratories, Altana, AstraZeneca, GlaxoSmithKline, Ivax, Merck, Sharpe and Dohme, Novartis, Schering Plough and Trinity Pharmaceuticals. He does not possess any pharmaceutical shares.
-
↵* A complete list of members of the COMPACT International Study Group is provided in the Appendix.
Linked Articles
- Airwaves