Article Text

Abstract
Over the last few decades attention has largely focused on airway inflammation in asthma, but more recently it has been appreciated that there are important structural airway changes which have been grouped together under the term “airway remodelling”. It is only now that questions have been asked about the impact of treatment on these structural changes. This review examines the nature of these structural airway changes, the mechanisms of their generation, their potential consequences, and what is known about the ability of anti-asthma treatments to modulate these changes.
- asthma
- airway remodelling
- pharmacotherapy
Statistics from Altmetric.com
Current understanding of asthma defines it as an inflammatory disorder of the airways involving T cells, mast cells, and eosinophils.1 While acute inflammation is a beneficial non-specific response of tissues to injury that generally leads to repair and restoration of normal structure and function, asthma represents a chronic inflammatory process of the airways followed by healing, whose end result is characteristically an altered structure referred to as a remodelling of the airways (see below). The possibility that these structural changes might be implicated in at least some of the clinical manifestations of asthma has been investigated in depth only in the last 10–15 years. However, despite extensive research, a number of important questions remain unanswered. The factors that initiate and perpetuate remodelling are incompletely understood. It is not clear whether airway remodelling is a normal response to an abnormal injury or, alternatively, whether the remodelling/repair response is itself an abnormal response. In addition, while remodelling is generally believed to be disadvantageous, in some ways it may serve a protective function. The balance between these effects in vivo is speculative. This review will outline the relevant morphological changes in the airway wall before discussing the possible consequences and reviewing what is known about the potential for pharmacotherapy to prevent or reverse these changes.
WHAT CONSTITUTES AIRWAY REMODELLING?
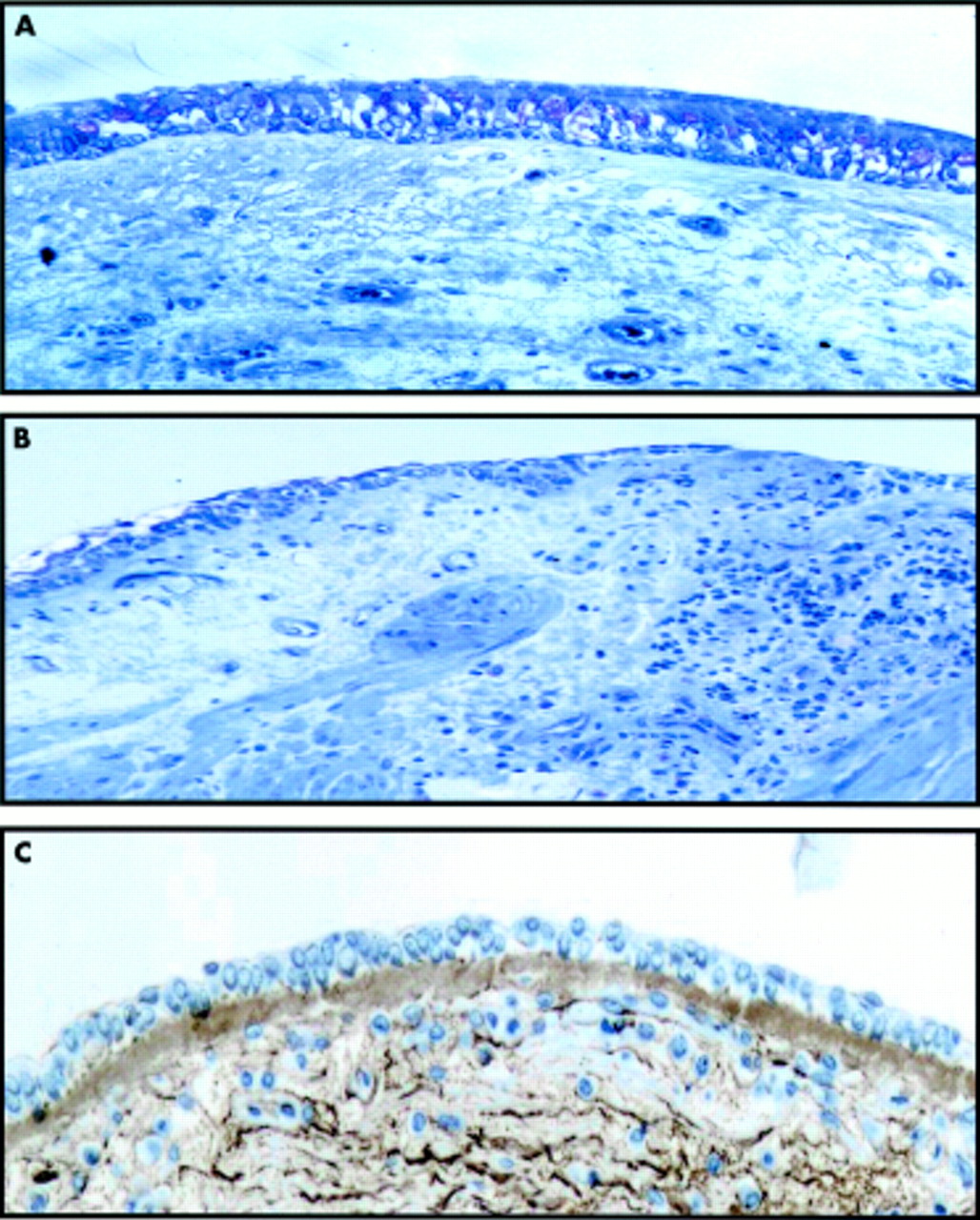
In addition to the inflammatory features mentioned earlier, the airway wall of patients with asthma is usually characterised by a number of structural changes that are grouped together under the umbrella term “airway remodelling”. It is not known which one(s) are the most useful and relevant markers, although most work has been done on epithelial disruption, sub-basement membrane thickening, and smooth muscle hypertrophy (fig 1). These changes are structural although, in the broadest sense, airway remodelling encompasses both structural and functional consequences of altered airway morphology.

Comparison of (A) normal and (B, C) asthmatic airway wall showing epithelial damage, increased smooth muscle, inflammatory cell infiltration, and sub-basement membrane thickening. The images in (A) and (B) have been stained with toluidine blue and the image in (C) has been stained with an antibody to collagen III.
Epithelial damage
Despite the variable preservation of airway epithelium in biopsy and necropsy specimens, careful sampling has revealed enhanced epithelial fragility and increased epithelial damage in asthma,2 and the extent of this abnormality correlates with measures of airway reactivity.3 This suggests that epithelial injury is not only a feature of asthma, but also appears to be related to asthma severity.
Sub-basement membrane thickening
Thickening of the lamina reticularis below the true basement membrane is a characteristic early feature of the asthmatic bronchus and has been termed “subepithelial fibrosis” or “sub-basement membrane thickening”. This thickening is associated with deposition of collagen I, collagen III, and fibronectin.4 An approximate twofold increase in sub-basement membrane thickness has been reported in asthmatics of varying severity.5,6 Interestingly, patients with rhinitis without asthma also show airway subepithelial fibrosis, although it is less marked than in asthma.7 In contrast, there is a lack of such thickening in patients with chronic obstructive pulmonary disease (COPD). The source of these matrix molecules is thought to be the subepithelial myofibroblast (α-smooth muscle actin positive) which forms a specialised network beneath the lamina reticularis.8 Workers have found a correlation between the thickness of the lamina reticularis and the number of subepithelial myofibroblasts.8–10 The origin of the myofibroblast remains unclear and is explored further in the section on mechanisms of airway remodelling.
Smooth muscle hypertrophy and hyperplasia
Increased smooth muscle mass within both large and small airway walls has long been recognised and is evident in both fatal and non-fatal cases of asthma.11,12 Necropsy studies suggest that there may be two patterns.13 In type 1 there is increased muscle mass associated with hyperplasia that is restricted to the large central airways; in type 2 there is mild hyperplasia in the large airways, but hypertrophy is detected throughout the bronchial tree especially in small peripheral airways. Increases in airway smooth muscle (ASM) mass may be due to several factors, including proliferation induced by inflammatory mediators, cytokines and growth factors; work hypertrophy due to repeated episodes of bronchospasm; reduced inhibitory control; and accumulation of enriched plasma in the environment surrounding the ASM. As well as changes in morphological aspects, some workers have described alterations in the functional characteristics of ASM in asthma. For example, it has been reported that asthmatic ASM has an increased maximal velocity of muscle shortening attributable to an increase in the activity of myosin light chain kinase,14 and this can explain the reduced bronchodilating effect of inspiration in asthmatics. Indeed, the loss of this protective mechanism has been suggested as one of the main causes of bronchial hyperresponsiveness (BHR).
Mucus metaplasia
Mucous glands are distributed throughout the airways in asthma and are even present in peripheral bronchioles where they are normally absent. The mucosal glands in the segmental bronchi of asthmatics are considerably enlarged, with a volume twice that of normal subjects.11,15 Increased goblet cells may be obscured by epithelial desquamation. One study, however, found no correlation between hyperplasia of mucous glands and asthma severity as evaluated by the degree of resting airflow obstruction.12
Increased vascularity
An increase in both the number and size of vessels has been reported in biopsy specimens from airways in asthmatic subjects compared with controls,16,17 although the relationship with asthma severity was not examined. This finding contrasts with studies comparing necropsy specimens from cases of fatal asthma, asthmatics who died of other causes, and non-asthmatics which do not reveal differences in total cross sectional vascular area, although in fatal asthma there is an alteration in the vascular distribution with an increase in the number and size of large vessels and a reduction in the number and size of small vessels.18 Enlarged congested mucosal blood vessels may contribute to the increased airway wall thickness described later. Examination of blood vessel distribution is obviously more difficult to evaluate in small superficial endobronchial biopsy specimens than in large necroscopic samples. The difference between these necroscopic findings and the in vivo biopsy studies may also relate to the method of vessel quantification. Staining for collagen IV appears to provide higher vessel counts than other markers of the endothelium.
Elastic fibre fragmentation
There are only a few studies and no consensus concerning the distribution and configuration of the fibres of the elastic system within the airways in asthma. Bousquet and colleagues reported fragmentation and paucity of fibres in the subepithelial layer in bronchial biopsy specimens of asthmatics of different severity.19 Mauad et al found elastosis and fragmentation of elastic fibres in central but not peripheral airways of cases of fatal asthma.20 However, Godfrey and co-workers could not find differences in the total content of elastic fibres in cases of mild, severe, or fatal asthma compared with controls.21
Extracellular matrix (ECM) glycoproteins
The ECM is composed of macromolecules such as polysaccharide glycosaminoglycans, proteoglycans, and fibrous proteins. In addition to the changes to collagen and elastin detailed above, there is enhanced fibronectin immunoreactivity4 and increased laminin-β2 deposition along the basement membrane,22 as well as increased expression of tenascin within the basement membrane.9,23 Proteoglycan deposition in the airway wall has been found to be significantly higher in asthmatic subjects than in controls, and in this study the degree of proteoglycan immunoreactivity was significantly correlated with airway responsiveness in the asthmatics.24
Airway nerves
Remodelling of neural components of the airway wall has been studied relatively little, but there is evidence for altered neural morphology and function in asthma. The only neural bronchodilator system in human airways is the inhibitory non-adrenergic, non-cholinergic (i-NANC) system. Putative neurotransmitters include vasoactive intestinal polypeptide (VIP) and substance P (SP). Ollerenshaw and colleagues showed that VIP was absent from the lungs of asthmatic subjects but was detected in 92% of non-asthmatic subjects.25 A number of studies have shown that the SP content of human airways is increased in asthma,26 while others have shown striking differences in the morphological characteristics of SP immunoreactive nerves in asthmatics.27 However, not all such studies have found alterations of VIP and SP in asthma.
MECHANISMS AND MEDIATORS OF AIRWAY REMODELLING
At its most basic level, remodelling has been attributed to the repetitive injury to the airway wall arising from cycles of inflammation and repair. With chronic persistent inflammation this is a continuous process. The asthmatic airway contains a vast array of inflammatory cells and mediators that have the capacity to affect profoundly the cellular and structural elements that are involved in remodelling. However, there is an indication that the remodelling process itself may be independent of inflammation, may be a primary event in the natural history of asthma, and may contribute to the development and persistence of the airway inflammatory process itself.
Much interest has focused on the transforming growth factor-beta (TGFβ) family of proteins. Many cells can produce TGFβ, but the important sources in the asthmatic airway are thought to be epithelium, fibroblasts, eosinophils, and airway macrophages. This growth factor is a potent profibrotic and anti-inflammatory cytokine which stimulates fibroblasts to promote the synthesis and secretion of many proteins of the ECM including collagen I, collagen III, fibronectin, vitronectin, tenascin, and proteoglycans.28–31 It also decreases the synthesis of enzymes that degrade the ECM, such as matrix metalloproteinases, and increases the synthesis of inhibitors of these enzymes, such as tissue inhibitor of metalloproteinase-1 (TIMP-1).32 TGFβ is capable of transforming fibroblasts into myofibroblasts and it is recognised that myofibroblasts have higher collagen synthesis activity than fibroblasts.33 It is reported to inhibit or increase proliferation of mesenchymal cells depending upon the concentration,34,35 is angiogenic in vivo, and is a potent chemoattractant for monocytes, neutrophils, T cells, and fibroblasts which, in turn, can elaborate inflammatory mediators. The potential importance of TGFβ in asthma is highlighted by in vivo studies showing altered compartmentalisation of TGFβ in asthmatic airways36 and increased levels in asthmatic bronchoalveolar lavage (BAL) fluid before and after antigen.37 In most studies TGFβ1 expression correlates with sub-basement membrane thickness and fibroblast number and/or disease severity.38–40 This may depend on whether it is evaluated at the mRNA level or the protein level, as much of the protein is complexed in an inactive form with airway proteoglycans.41
Modulating the effects of profibrotic growth factors are the matrix metalloproteinases (MMPs). These endopeptidases are produced by tissue structural cells including fibroblasts and endothelial cells, and by inflammatory cells such as macrophages, neutrophils, and eosinophils. The production and action of the MMPs are subject to strict control, ensuring limited proteolysis of the ECM during cell migration, tissue remodelling and repair, and are inhibited by a group of specific inhibitors—the tissue inhibitors of metalloproteinases (TIMPs).42,43 Inadequate regulation of MMP production or action has been implicated in a wide range of pathological processes including rheumatoid arthritis, renal diseases, osteoarthritis, fibrotic disorders, and tumour invasion.44 Studies have suggested an imbalance between MMPs and TIMPs in the asthmatic airway, perhaps favouring the development of fibrosis.45
Epidermal growth factor (EGF) and its receptor (EGFR) may also play a key role in airway remodelling and repair.46 EGF is expressed at high levels by epithelial cells, endothelial cells, macrophages, and platelets; it participates in the repair processes involved in resurfacing of a wound with new epithelium and promotes healing by stimulating proliferation and migration of epithelial cells and increasing the synthesis of proteins such as fibronectin. As damaged epithelium can secrete TGFβ and this growth factor can inhibit the epithelial repair process, the balance between EGF and TGFβ potentially determines the state of epithelial repair and the availability of growth factors to modify the ECM. In addition, EGF can act as an angiogenic factor by stimulating DNA synthesis of endothelial cells, as well as their migration and proliferation. Both EGF and EGFR expression are found in increased quantities in asthmatic airways.39,47 Vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF) are two other growth factors relevant to the angiogenic processes within the airways, and FGF—which has increased recovery in the BAL fluid of patients with asthma48—has also been linked to fibroproliferative processes. Expression of VEGF and its receptors is upregulated in asthma, and the degree of airway vascularity has been found to correlate with this expression.49 Other potentially important mediators abound, and in many cases increased expression of these mediators has been seen in asthmatics, including granulocyte macrophage-colony stimulating factor (GM-CSF),39 tumour necrosis factor-α (TNF-α),50 endothelin,51 tryptases,52 thrombin,53 histamine, and leukotrienes.54 Finally, work on transgenic mice has indicated a possible role for interleukin (IL)-6,55 IL-11,56 IL-13,57 IL-5, and IL-958; overexpression of all these cytokines in the airway leads to morphological changes reminiscent of asthmatic airway remodelling.
Although inflammation per se may be responsible for part of the remodelling response, it seems that the bronchial epithelium is especially important in orchestrating repair and proliferative responses that interact with the inflammatory component. As a result of an impaired response to injury, the epithelium becomes a major source of growth factors, the action of which on underlying structural cells could explain the fibrotic responses. The concept of the reactivation of the epithelial-mesenchymal trophic unit (EMTU) has emerged from these observations.59 The network of myofibroblasts beneath the lamina reticularis forms a so-called “attenuated fibroblast sheath” close to the epithelium,60 giving rise to an integrated unit that is capable of responding to exogenous and endogenous stimuli. The EMTU is so named because of its pivotal role in fetal lung development where the release of soluble mediators by epithelial cells and myofibroblasts is crucial at different times of airway growth and branching.61 Work on in vitro co-culture models has shown that blocking EGFR mediated bronchial epithelial repair leads to augmented release of pro-fibrogenic growth factors62 which enhanced collagen gene expression when applied to cultures of bronchial myofibroblasts. In turn, production of growth factors from fibroblasts might encourage proliferation of fibroblasts, smooth muscle cells, and blood vessels. Production of other factors by fibroblasts has the potential to amplify the inflammatory process—for example, by the action of IL-6 on T cells, GM-CSF on eosinophils, stem cell factor on mast cells, and eotaxin on mast cells and eosinophils. Likewise, it has recently been appreciated that smooth muscle cells have the capacity to secrete such cytokines and growth factors.63 In this way, signals from the epithelium are propagated and amplified throughout the airway wall. Mesenchymal derived mediators (such as TGFβ) may also signal upwards through the airway wall to act on the bronchial epithelium where they may antagonise epithelial repair processes (such as those driven by EGF).
WHAT ARE THE CONSEQUENCES OF AIRWAY REMODELLING?
Effects on airway inflammation
Although remodelling has been considered a consequence of inflammation, there is an indication that the remodelling process itself may be independent of inflammation, may be a primary event in the natural history of asthma, and may contribute to the development and persistence of the airway inflammatory process itself. In this respect, work showing that ECM components such as fibronectin can interact with inflammatory cells, influencing cell activation, mediator release and chemotaxis, is of great interest. It has also been postulated that trafficking of dendritic cells—the primary antigen presenting cell in the airways—may be hampered by changes in the ECM leading to an enhanced immune response while still in the airway wall.
Effects on airflow obstruction
For many years asthma has been considered to be a condition characterised by airflow obstruction that is reversible, either spontaneously or in response to treatment. However, cross sectional studies have shown that many asthmatics—both symptomatic64–66 and asymptomatic67—have evidence of persistent irreversible airflow obstruction. In addition, studies have confirmed that asthmatics as a group experience an accelerated rate of deterioration in respiratory function.68–70 For example, Peat and co-workers69 in Australia found a rate of decline in forced expiratory volume in 1 second (FEV1) of 50 ml/year in asthmatic non-smokers and 35 ml/year in controls. A more recent community based survey over 15 years, the Copenhagen Heart Study, also collected lung function data and found values of 38 ml/year and 22 ml/year, respectively.70 In both of these studies smoking had an additional effect, with asthmatic subjects who smoked having an even greater accelerated fall in lung function with time than asthmatic non-smokers (fig 2).

{kind=link}
{kind=link}
Changes with age in height adjusted forced expiratory volume in 1 second (FEV1) according to sex, smoking status, and the presence or absence of asthma. The curves in each panel differ significantly from each other (p<0.001). Reproduced with permission from Lange et al.70
It is tempting to ascribe this irreversible and accelerated loss of lung function to the structural airway changes of remodelling. Subepithelial fibrosis has been most studied in this regard, mainly because of the ease with which it can be measured in bronchial biopsy specimens. Some studies have found a correlation between FEV1 and sub-basement membrane thickening,9,38,71 but others have failed to find such a correlation.4,72 The timing of FEV1 measurement is important since post-bronchodilator FEV1 may be a better measure than resting pre-bronchodilator FEV1 in terms of the structural airway changes. In fact, histopathological studies have shown that sub-basement membrane thickening may be seen in newly diagnosed asthmatic patients.73 If basement membrane thickening can be found in newly diagnosed asthmatic patients and persistent obstruction is correlated with disease duration, as reported by some investigators,64,65 then subepithelial fibrosis would appear to have a limited role in irreversible airflow obstruction and perhaps smooth muscle changes are more relevant. Such measures are, however, made in samples from large airways and there is little information in vivo on fibrotic change in the small airways. These changes are evident in resected/necroscopic samples and could well account for the loss of bronchodilator response and impaired resting lung function in asthma.
Effects on bronchial hyperresponsiveness (BHR)
Bronchial hyperresponsiveness to non-specific stimuli is a characteristic feature of the asthmatic patient. While a number of mechanisms, including epithelial desquamation, may contribute to this enhanced responsiveness, it has been proposed that structural airway changes, particularly those associated with airway wall thickening, are a substantial determinant of this physiological abnormality. The net result of many of the morphological changes described above—particularly increased smooth muscle and collagen deposition in the airway wall—is to increase the overall thickness of the airway wall, a finding highlighted as long ago as 1922 by Huber and Koessler.74 Using mathematical models it has been shown that, although there may be little effect when the ASM is relaxed, the wall thickening can enhance the degree of luminal narrowing caused by a given degree of muscle contraction.75–78 In addition, subepithelial collagen deposition could increase the stiffness of the inner layer of the airway wall in comparison with the surrounding submucosa, a phenomenon that would be expected to lead to the formation of a small number of large mucosal folds instead of a large number of thin folds when the airway buckles in response to smooth muscle contraction.79 The consequence is again a larger increase in the intraluminal airway pressure in response to the same degree of smooth muscle contraction. Finally, increased thickness of the outer wall could uncouple the airway from the distending and tethering forces of surrounding lung parenchyma, an effect that would diminish the effects of increasing lung volumes on increasing airway diameter.80
Consistent with the premise that BHR is a marker of airway remodelling is the finding of a correlation between subepithelial fibrosis and BHR in several,5,12,71,72 but not all4 studies. This discordance may be related to the narrow range of severity studied because of the invasive nature of bronchoscopy, and to variations in treatment taken by subjects. In addition, it has been suggested that it is the density rather than the thickness of the lamina reticularis that is the most relevant measurement. Similarly, increased deposition of proteoglycans in the airway wall in asthmatic subjects has been reported to correlate with BHR.81 Further evidence that BHR may provide a surrogate physiological marker for airway wall remodelling comes from studies that have shown a relationship between BHR and the annual decline in FEV1.69,82 However, the significance of a correlation between sub-basement membrane thickness and BHR is not clear. The correlation itself does not imply a causal relation because both findings may be unrelated consequences of airway inflammation.
Harmful or protective?
It is important to appreciate that functional consequences might not always be deleterious. For example, in a rat model exposure to aerosolised ovalbumin (OA) results in increased airway wall thickness reminiscent of asthmatic remodelling.83 Such rats initially develop increased airway responsiveness but, with continued exposure, scarring occurs with increased collagen deposition beneath the basement membrane and a consequent reduction in the thickness of the airway wall. Concurrently, the hyperresponsiveness improves and may even develop into a hyporesponsiveness. However, these animal models do tend to show more widespread fibrosis than that seen in human asthma. Equally, increased collagen deposition around the smooth muscle layer may interfere with smooth muscle contraction, thus effectively protecting against bronchoconstriction.84 It is likely that such fibrosis of the airway wall would replace the variability in expiratory flow characteristic of asthma for a fixed permanent element of airflow obstruction more reminiscent of COPD, and this could be considered a protective response. We should be careful to keep these contradictions in mind whenever we make attempts to prevent or reverse remodelling.
EFFECT OF ANTI-ASTHMA TREATMENT ON REMODELLING
Glucocorticosteroids
Glucocorticosteroids (hereafter termed “steroids”) have revolutionised the treatment of asthma and have now become the mainstay of therapy, being recommended in national and international guidelines.85,86 Although steroids have a broad range of effects, the important effects with regard to asthma relate to their ability to inhibit inflammatory processes. Discussion of the details of the anti-inflammatory effects of steroids is beyond the scope of this article but is reviewed elsewhere.87
As remodelling of the airway wall is thought to be a result of chronic inflammation within the bronchial wall, it follows that, since steroids reduce or reverse inflammation, they may also reduce or reverse remodelling. However, as well as an indirect effect on remodelling through the modification of inflammation, there is also in vitro evidence for a direct effect of steroids on the cells, growth factors, and cytokines thought to be central to the remodelling process. Some of the relevant experimental evidence is shown in box 1.
Box 1 In vitro effects of corticosteroids on remodelling processes
-
Increased or decreased smooth muscle fibronectin production, depending on steroid92
-
Decreased fibroblast TGFβ expression95
-
Increased expression of SLPI (secretory leukocyte protease inhibitor) by epithelial cells105
-
Decreased VEGF expression by epithelial cells106
-
Reduced adhesion molecule expression by endothelial cells, fibroblasts, and epithelial cells107–109
-
Reconstitution of epithelial structure110
-
Decreased mucus production111
-
Decreased expression of cytokines and chemokines by a variety of cells87
Little is known about the effect of steroids on collagen production by asthmatic airway fibroblasts. While in vitro observations would suggest a prominent role for steroids in influencing airway remodelling, in vivo studies on non-invasive measurements (airflow obstruction and BHR) and invasive measurements (mainly lamina reticularis thickness) have provided conflicting results with no consensus as to an effect on the remodelling per se. The impact of treatment on airflow obstruction can be assessed both in terms of reversibility and in terms of maintaining improved lung function and preventing an exaggerated decline in lung function with time. Steroids do not have a rapid bronchodilator effect, but lead to a progressive improvement in lung function over weeks to months.112–114 These changes, however, can all be explained by the anti-inflammatory effects of treatment on the airway and do not provide information as to whether these agents modify the remodelling process itself; such information is best evaluated by long term studies.
Perhaps surprisingly, there are very few studies addressing the long term impact of treatment on the rate of decline in lung function, probably because it would be considered unethical to establish a long term study in which there was an untreated control population. The limited information available has therefore either come from early studies comparing steroids with β agonists, from non-controlled comparisons of groups on different treatments, or from follow up of patients within one treatment group. There is evidence that the annual decline in lung function may be slowed by the introduction of inhaled steroids.115 Increasing evidence also suggests that delay in starting inhaled steroids may result in less overall improvement in lung function in both children and adults. A relevant study compared inhaled steroids as first line treatment against β2 agonist alone.112,116 Adult patients with asthmatic symptoms of less than 1 year and no previous use of anti-inflammatory treatment were randomised to receive budesonide or terbutaline as the first and only regular medication. Two years of treatment with budesonide resulted in almost complete clinical recovery and normalisation of lung function, and was superior to terbutaline. The study was continued for a third year to investigate the effects of discontinuation and delayed introduction of inhaled steroids. Most patients who switched from budesonide to placebo showed a gradual and slight decline in lung function, but 20% did not deteriorate at all. Those patients who switched from terbutaline to budesonide improved but did not reach the same level of lung function or improvement in bronchial responsiveness as those who were treated with inhaled steroids initially. Consistent with this is a retrospective analysis by Agertoft and Pedersen117 of children who, following the diagnosis of asthma, were treated either with steroids or with other therapies (including chromones, theophyllines and regular β2 agonists).117 The non-steroid treated children who were subsequently transferred to inhaled steroid therapy had a reduced annual improvement in % predicted FEV1 as they grew compared with the primary steroid treated group. A further retrospective analysis from Finland of adults with asthma evaluated the correlation between the response to treatment with inhaled steroids and the duration of symptoms before starting inhaled steroids for the first time.118 A negative correlation between improvement in lung function following inhaled steroids and duration of asthma was found.
All these reports give persuasive evidence that early treatment with inhaled steroids may prevent the development of airway events—presumed to be those associated with structural remodelling—that lead to irreversibility in lung function, but do not reverse these changes once they have arisen. Such a conclusion is supported by a more recent prospective study of 101 asthmatic subjects who were studied at age 5–14 years, 22–32 years, and 32–42 years. Subjects who continued to use inhaled steroids had a significantly smaller annual decline in FEV1 from visit 2 to visit 3, adjusted for level of attained FEV1 at visit 2.119 However, the presence in the literature of conflicting reports of decline in lung function in asthma despite steroid treatment64,66,120 might suggest that the protective effect of steroids is only partial or that it may require a higher dose than that which is usually employed to see a consistent effect in all patients. Support for this comes from the recent CAMP study in which the initial benefit of inhaled steroids on FEV1 during the first year of treatment was lost over the following 3 years.121
Steroid treatment leads to rapid improvements in histamine and methacholine bronchial reactivity that can be explained by their anti-inflammatory effect and do not necessarily indicate an effect on remodelling.112,122 One study compared the effects of inhaled budesonide and oral prednisolone and found that, while both treatments improved lung function, only the inhaled steroid improved airway reactivity.123 This suggests the importance of topical activity in the improvement of BHR. One study has shown that both delayed and early introduction of inhaled steroids resulted in similar increases in FEV1, although early introduction resulted in greater improvement in bronchial responsiveness to histamine.124 However, studies in both children and adults112,113,125 have shown that prolonged treatment can result in a further progressive improvement in BHR that does not plateau until 9–18 months of treatment. In contrast to lung function measures which improve over 2–4 weeks, this progressive improvement would be consistent with a gradual reduction in the airway remodelling. In patients with mild asthma this improvement in reactivity is associated with a limitation in the maximum airway narrowing achievable with bronchoconstrictor stimuli such as methacholine, indicative of a fundamental change in airway behaviour. It is noteworthy that, although BHR improves in most long term studies, it does not return to normal limits.126 Although several potential mechanisms exist for the improvement in BHR, it is probable that, while the rapid improvement seen over the first month may relate to a reduction in inflammation and a decrease in epithelial activation, the longer term gradual improvement is likely to relate to resolution of airway remodelling. Consistent with this, it has been shown that asthma rapidly regresses within 1 week of stopping inhaled steroid treatment of short duration (6 weeks), but there is long lasting improvement (>3 months) after continuous treatment for 1 year.116,127,128
The final arbiter as to whether treatment modifies the airway wall remodelling process comes from direct assessment of airway events before and after treatment. It is beyond argument that inhaled steroids cause a reduction in the cellular infiltrate of bronchial biopsy specimens usually resulting from decreased eosinophils, mast cells, and lymphocytes.122 Similarly, it has been reported that corticosteroid treatment improves epithelial morphology, returning the disrupted appearance toward an intact, normal looking epithelium.129,130 A number of studies have shown that inhaled steroids are able to reverse airway collagen deposition and reduce the thickness of the subepithelial basement membrane,10,131–134 but several have reported no effect.129,135 The AMPUL study group showed that inhaled steroids reduced the thickness of the subepithelial basement membrane, but to a greater extent if treatment was additionally guided by measurement of bronchial responsiveness.134 One study has shown a reduction in tenascin in the subepithelial basement membrane in pollen sensitive patients treated with beclomethasone 400 μg twice daily compared with placebo,23 and another has shown that inhaled steroids decrease MMP-9 and increase TIMP-1 expression in asthmatic bronchial mucosa.10 Hoshino and colleagues have also shown that inhaled steroids cause a reduction in IGF-1 expression in bronchial mucosa which is associated with reduced subepithelial basement membrane thickness although, surprisingly, there was no change in TGFβ1 or PDGF expression.133 The same group has reported a reduction in airway wall vascularity following treatment with inhaled steroids.136 A valid criticism of most studies is the use of relatively short treatment periods using only modest doses of inhaled steroids. It may also be less possible to reverse remodelling once it is established rather than preventing its development in the first place. For example, it has been shown that patients with both recently diagnosed and long standing asthma have similar degrees of airway inflammation and subepithelial fibrosis73 and, although BHR could be improved, it could not be normalised in either group. This suggests that, even in patients with asthma of recent onset, there are irreversible airway changes. This concept is supported by the knowledge that there is subclinical inflammation and remodelling in the absence of frank asthma, and by animal models where remodelling in OA sensitised rodents could not be reversed but was partly prevented by concomitant treatment with steroids during the allergen exposure.137,138 As already indicated, subepithelial fibrosis represents only one aspect of remodelling and, because of the difficulties involved in tissue sampling, it is unclear whether other structural changes that contribute to airway wall thickening are of greater relevance.
β2 agonists
β2 agonists act via binding to the β2 adrenergic receptor, a transmembrane receptor that is coupled to G proteins. Receptor activation increases intracellular cAMP which, by decreasing intracellular calcium, leads to smooth muscle relaxation.139 While β2 agonists are primarily considered simply as bronchodilators, it is possible that prolonged β2 receptor stimulation could influence some of the pathways contributing to airway inflammation and remodelling in asthma. Apart from smooth muscle, β2 adrenoceptors are also expressed on many other cells types in the airway and research has shown that β2 agonists may have pharmacological properties useful for controlling inflammation.140 For example, in vitro studies have shown decreased plasma exudation141 as well as inhibition of mediator release from smooth muscle cells,93,94 mast cells,142 eosinophils,143 monocytes,144 and lymphocytes.145 A number of studies have reported inhibition of smooth muscle cell146,147 and fibroblast proliferation148 by β2 agonists. The mechanism whereby β2 agonists exert these effects is unclear, but it is known that cAMP can activate protein kinase A which is able to phosphorylate an important nuclear transcription factor called cAMP response element binding protein (CREB). Phosphorylated CREB is able to bind to segments of DNA and can thus influence gene transcription, with the effects being dependent on the type of cell involved.
Despite these promising in vitro findings, there is little evidence for an in vivo effect on remodelling when β2 agonists are used as sole treatment. Understandably, bronchodilator drugs improve lung function in the short term, but their effects are limited to the duration of action of the drug within the airway. Cessation of treatment leads to a rapid decline in lung function, indicating that these protective effects are due to functional antagonism of bronchoconstriction and are not related to any fundamental effect on airway structure. Similarly, any drug that relaxes the ASM will functionally antagonise constrictor stimuli and will thus modify bronchial responsiveness during the period of pharmacological activity149,150; however, there does not appear to be any fundamental effect on airway reactivity in the long term.
In contrast to studies showing increased airway inflammation during monotherapy with short acting β2 agonists,151 a few clinical studies have shown evidence of anti-inflammatory effects of long acting β2 agonists.152–156 However, several clinical studies failed to show any effect150,157 and, at the present time, only one study has reported positive effects of β agonist therapy on remodelling.16
Theophyllines
Theophylline has been used for decades both as a bronchodilator and a respiratory stimulant. The mode of its bronchodilator action is thought to be relaxation of bronchial smooth muscle by inhibition of phosphodiesterase (PDE), which breaks down intracellular cAMP. As with the β2 agonists, there is a growing recognition that theophylline may act in ways other than PDE inhibition and may have anti-asthma effects other than bronchodilation, with several anti-inflammatory effects occurring at concentrations that are therapeutically relevant158,159—for example, it has been reported that theophylline decreases mediator release from mast cells, monocytes, and T lymphocytes. Alongside these potential effects on inflammation are reports that theophylline is able to inhibit fibroblast proliferation.160
As for β2 agonists, the effects of theophylline on lung function and BHR are short term and reverse after discontinuation of the drug. Although theophylline protects poorly against the early asthmatic response following allergen challenge, most studies show a marked protective effect against the late response. The former is associated with bronchospasm while the latter is associated with inflammation, so this observation suggests that theophylline is acting in a manner other than as a smooth muscle relaxant.161,162 It was shown that theophylline practically abolished the late asthmatic reaction to inhaled allergen and that this was associated with a reduction of increases in CD4+ and CD8+ T cells in peripheral blood.163 Sullivan et al164 reported that bronchial biopsy specimens taken 24 hours after allergen challenge revealed a significant reduction in total and activated eosinophils in the airway mucosa of patients who received theophylline compared to placebo. In a study of the effects of discontinuing theophylline in patients already on high dose inhaled steroids, there was an increase in the number of CD4+ and CD8+ cells and activated eosinophils in the bronchial wall.165 Similarly, in a group of patients in whom theophylline was added to inhaled steroids the number of CD8+ cells and the number of cells containing IL-4 and IL-5 were reduced.166 However, despite the evidence for an anti-inflammatory effect, there have been no long term studies of the effect of theophylline on remodelling.
Chromones
Chromones are thought to act by stabilisation of the mast cell membrane, thus inhibiting mediator release; however, doubts have been expressed about this mechanism of action and it may be that interaction with sensory nerves or other inflammatory cells is more important. There is no evidence that long term treatment fundamentally alters lung function or airway reactivity outside the period that the drug is pharmacologically active. While chromones protect against laboratory allergen challenge induced increments in airway reactivity167 and seasonal increases in bronchial responsiveness in pollen sensitive subjects,168 this is probably linked to airway wall oedema and inflammatory cell influx rather than structural changes. Direct evidence is thus needed to clarify whether chromones prevent or modify such structural events. The effects of treatment on biopsy169 and BAL fluid170 cell counts have been conflicting and a reduction in cell activation rather than cell recruitment may be more relevant to the effect of chromones. Despite the modest evidence for an anti-inflammatory effect, however, there have been no long term studies of the effect of chromones on remodelling.
Leukotriene modifiers
The 5-lipoxygenase pathway is the name given to the series of biochemical reactions which result in the transformation of arachidonic acid (present in cell membranes) into leukotrienes. Leukotrienes can induce many of the abnormalities seen in both acute and chronic asthma, including bronchoconstriction, increased permeability of vessels, mucous gland secretion, proliferation of the ASM, and inflammatory cell infiltration. Studies in vitro have shown that LTD4 can induce smooth muscle hyperplasia in the presence of EGF or PDGF, so it is theoretically possible that inhibition of leukotriene activity might lead to a reduction in smooth muscle remodelling.171,172 Leukotriene modifiers were introduced into clinical practice in 1995 but, to date, there have been no long term intervention studies to evaluate fully the effects of this class of drug on long term changes in lung function. Similarly, there is little evidence for a concerted effect of leukotriene receptor antagonists on bronchial reactivity, apart from work presented as abstracts.173,174 There is only one published report of an improvement, a 1.87-fold shift in the threshold dose of methacholine inducing bronchoconstriction over a 4 week period with oral pranlukast.175 Although it is possible that a more consistent and greater effect would be seen with more prolonged treatment, any effect on BHR is unlikely to represent an effect on remodelling as it has been shown that LTC4 and LTD4 can themselves increase BHR to methacholine or histamine.176 There is only one published bronchial biopsy study, in which a reduction in T cells, mast cells, and activated eosinophils in the airway was reported after 4 weeks of treatment of asthmatic subjects with pranlukast compared with placebo.175 As would be expected for this newly introduced class of drugs, no studies have been undertaken to look at the effects on airway remodelling.
Immunosuppressive therapy
Immunosuppressive therapy (methotrexate, cyclosporin) has been used in patients with severe asthma when other treatments are unsuccessful or in an attempt to reduce the dose of oral steroids required. While these drugs have powerful effects on immune and inflammatory pathways, they also have common and potentially serious side effects. For this reason, no studies have been carried out on their effects on airway remodelling and it is very unlikely that such a study would ever be contemplated.
Combination therapy
A number of clinical studies have shown that addition of a long acting β2 agonist to a low dose of inhaled corticosteroid gives a better outcome than doubling the dose of the corticosteroid in terms of a number of end points, including exacerbation rate.177,178 As a result, combination therapy is now recommended and widely prescribed for asthmatics at and above stage 3 of the BTS asthma guidelines.85 It is therefore valid to consider whether such combination therapy might affect remodelling. Although the benefits of such combination therapy may partly reflect the different basic pharmacological effects (that is, anti-inflammatory for steroids, bronchodilation for β agonists), this does not explain why the addition of short acting β agonists to steroids has a limited benefit. Attention has recently focused on the possible interactions at a cellular level between these drugs.
Glucocorticosteroid molecules act by passing through the cell membrane and binding to specific cytoplasmic receptors, the glucocorticoid receptor (GR). The activated GR produces its effects by regulating the transcription of target genes.87 As already discussed, β2 agonists act through the β2-AR and, in addition to the effects on smooth muscle, they activate protein kinase A. This enzyme is able to phosphorylate CREB, an important co-factor affecting gene transcription and so, through this pathway, gene transcription may be altered by β agonists. With these pathways in mind, there have been several mechanisms described whereby β agonists and steroids might interact (box 2).
Box 2 Possible mechanisms of interaction between β agonists and steroids
-
β receptor stimulation leads to a downregulation of responses to subsequent stimulation. Studies have shown that this desensitisation can be reversed by exposure to corticosteroids.179
-
There is indirect evidence that β agonists might influence glucocorticoid receptor (GR) function. Forskolin which, like β agonists, increases intracellular cAMP, activates GR transcription,180 increases cellular GR numbers,181 and may antagonise the downregulation of GRs induced by dexamethasone.182
-
So-called “cross-talk” between transcription factors such as the GR, CREB, NFκB, and AP-1 has also been described—for example, β2 agonists, through the action of protein kinase A, are able to phosphorylate cAMP response element binding protein (CREB), and this phosphorylation can inhibit activation of CREB binding protein (CBP) by glucocorticoid receptors.183 This effect would be expected to lead to a reduction in the efficacy of steroids and was suggested as a mechanism for the possible adverse effects of short acting β2 agonist therapy on asthma control. However, in a clinical study there was no evidence of a negative interaction between terbutaline and budesonide in the DNA binding of NFκB, GR, and CREB in bronchial mucosa.184 As other forms of interaction have been appreciated, it has been realised that this inhibitory form of interaction is not dominant. For example, β2 adrenoceptor induced protein kinase A (PKA) activation can switch coupling of the β2 adrenoceptor from the Gs (stimulatory) to the Gi (inhibitory) protein, thereby activating the mitogen activated protein kinase (MAPK) second messenger system.185 This results in induction instead of inhibition of CBP activation.186
-
Eickelberg and colleagues187 have shown that β2 agonists are themselves able to activate GR, leading to its translocation from the cytosolic to the nuclear compartment.
In vitro studies have shown functionally relevant interactions between β2 agonists and corticosteroids. A synergistic inhibition of TNF-α stimulated IL-8,93 and eotaxin release from human ASM cells has been reported.94 Similarly, GM-CSF release from TNF-α stimulated human epithelial cells was reduced by approximately 50% by budesonide but not by formoterol, although when both drugs were used a synergistic reduction in GM-CSF release of 75% was observed.188 More recently, formoterol was shown to have an additive effect over and above that of budesonide on IL-1β induced expression of adhesion molecules by fibroblasts.108 Clinical studies have added weight to these in vitro observations. Although some studies have reported increased airway inflammation during monotherapy with short acting β2 agonists,151 this has not proved to be the case with long acting β2 agonists. Indeed, one study has reported a reduction in submucosal mast cells and IL-4 positive cells with combination therapy,189 while Ward and colleagues have shown reduced levels of IL-8 in BAL fluid.190 In terms of remodelling, increased airway wall vasculature, a feature of chronic asthma, may be reduced by combination therapy.16 Further studies with large numbers of patients are needed to investigate whether combination therapy truly has important effects on remodelling.
CONCLUSIONS
Inhaled corticosteroids remain the class of drugs most likely to affect remodelling, but it is likely that they will need to be started early in the course of the disease. However, even those patients whose asthma responds to steroid treatment have persistent abnormal airway reactivity and some subjects have a very poor response to steroids. The possibility of other therapies such as β2 agonists enhancing the effect of steroids offers another exciting avenue of investigation (although as yet any interactive effects in vitro still need to be proved in vivo), as does the development of new treatments. Effective treatment of airway remodelling may require the development of novel therapies directed against cells or mediators that are key to the development of remodelling. There are a large number of possible targets, such as TGFβ, PDGF, bFGF, IGF, TNFα, MMPs, interleukins, and endothelins. For example, in a mouse model of atopic asthma, Blyth and colleagues were able to show that treatment with an anti-IL-5 antibody was able to limit the development of subepithelial fibrosis.191 Similarly, recent work showing the association of the ADAM33 gene with both asthma and bronchial hyperresponsiveness192 suggests that this class of protein may be an important therapeutic target. As these new therapies become available—as they undoubtedly will—it is important that prospective, longer term clinical studies are undertaken with end points other than just symptoms and peak flow—for example, bronchial reactivity and rate of decline of FEV1—which may give insight into the impact of treatment in the remodelling process. In addition, improved methods of imaging the airways to monitor structure would be of obvious value. In this respect, advances in imaging techniques such as CT scanning or endobronchial ultrasound may prove useful in the future. In addition, it is likely that no single treatment will effectively inhibit all aspects of remodelling, and that greater consideration may be needed as to the specificity of the target of individual treatments. For the present, further in vitro and in vivo work needs to be carried out to characterise more clearly the effects of anti-asthma drugs on airway remodelling processes. For the future, improved understanding of the pathophysiology of remodelling holds the promise of new and more effective anti-asthma treatments.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵