Article Text

Abstract
We present a case of bronchocentric granulomatosis in a woman with no history of asthma who was colonised with Aspergillusfumigatus. A family history of chronic granulomatous disease prompted further testing that demonstrated severely depressed neutrophil oxidant production and gp91phox deficiency compatible with the X linked carrier state of chronic granulomatous disease. Only one report of the association of these two rare diseases has previously appeared in the literature. We postulate that an ineffective immune response led to the prolonged colonisation of Afumigatus resulting in a hypersensitivity reaction that was manifest clinically as bronchocentric granulomatosis.
- chronic granulomatous disease
- bronchocentric granulomatosis
- Aspergillus
Statistics from Altmetric.com
Bronchocentric granulomatosis (BCG), first described by Liebow in 1972, is characterised by inflammation that begins within and is centred around bronchiole walls.1 Katzenstein et al described two patterns of disease.2 The more common form occurs in asthmatic patients and the prevalence appears to be increased in young men. It manifests with fever, cough, wheezing, dyspnoea, and blood eosinophilia. The radiographic patterns include bilateral airspace disease and a macronodular or, rarely, a reticulonodular interstitial pattern. In non-asthmatic patients BCG tends to occur at an older age and is generally less severe. Eosinophilia is uncommon but the chest radiographic findings are similar to those in asthmatic patients.
We report a case of biopsy proven BCG in a woman with no prior or subsequent history of asthma who was discovered to be a carrier of X-linked chronic granulomatous disease with skewed inactivation of one of her X chromosomes. We propose a hypothesis linking these two rare conditions.
CASE HISTORY
A 41 year old woman presented with a severe non-productive cough, fevers, anorexia, and myalgias. Over the preceding 2 weeks she had been admitted to hospital twice for progressive dyspnoea. Laboratory investigations showed an increased white cell count of 27.4 × 109/l with 11% eosinophils, and an ESR of 94. The chest radiograph revealed a new diffuse, predominantly reticulonodular, interstitial pattern. Her past medical history was significant for a clinical diagnosis of ulcerative colitis 13 and 3 years earlier, a skin rash that was compatible with cutaneous lupus erythematosus on biopsy 2 years earlier, and recurrent infections including frequent episodes of acute cervical adenopathy, sinusitis, a root canal abscess, and a slowly resolving right middle lobe pneumonia 5 months earlier. She had a 20 pack year smoking history. There was no prior history suggestive of asthma or atopy. Her family history was significant for a brother who had died at the age of 4 from the consequences of immunodeficiency. On examination she appeared chronically unwell and was in mild respiratory distress. Her oxygen saturation was 91% on room air and few bibasilar inspiratory crackles were present.
A high resolution CT scan of the chest showed a diffuse small nodular interstitial pattern with cystic changes at the right apex but no bronchiectasis. Pulmonary function tests revealed normal lung volumes, flows and transfer factor. Serological tests for ANA, RF, ANCA, and HIV antibodies were negative. Serum IgE was significantly increased at 9000 IU/ml (normal <100). Serum precipitins for Aspergillus fumigatus were positive using an immunodiffusion technique. Sputum cultures grew Afumigatus. Bronchoscopic examination with transbronchial lung biopsy showed non-specific chronic inflammation. The bronchoalveolar lavage cultures were negative for bacteria, mycobacteria, and fungi, and a cytological examination was unremarkable. Further history revealed several possible sources of exposure to Aspergillus spores including bird droppings found in the central air vent of her house 2 months before and mouldy wood chips used in her garden 1 month earlier.
Over the week following her transfer she had increased shortness of breath. Repeat pulmonary function tests showed severe restriction (total lung capacity (TLC) 60% predicted, forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) 47% predicted, carbon monoxide transfer factor (Tlco) 59% predicted). She underwent an open lung biopsy of the lingula which showed extensive panmural bronchocentric necrotising granulomatous inflammation with increased numbers of eosinophils in the airways (fig 1). Silver methenamine stain revealed occasional degenerated fungal elements consistent with Aspergillus within the affected airways but no evidence of tissue invasion. Other stains including Ziehl-Neelsen were negative. The pathology was diagnostic of BCG.
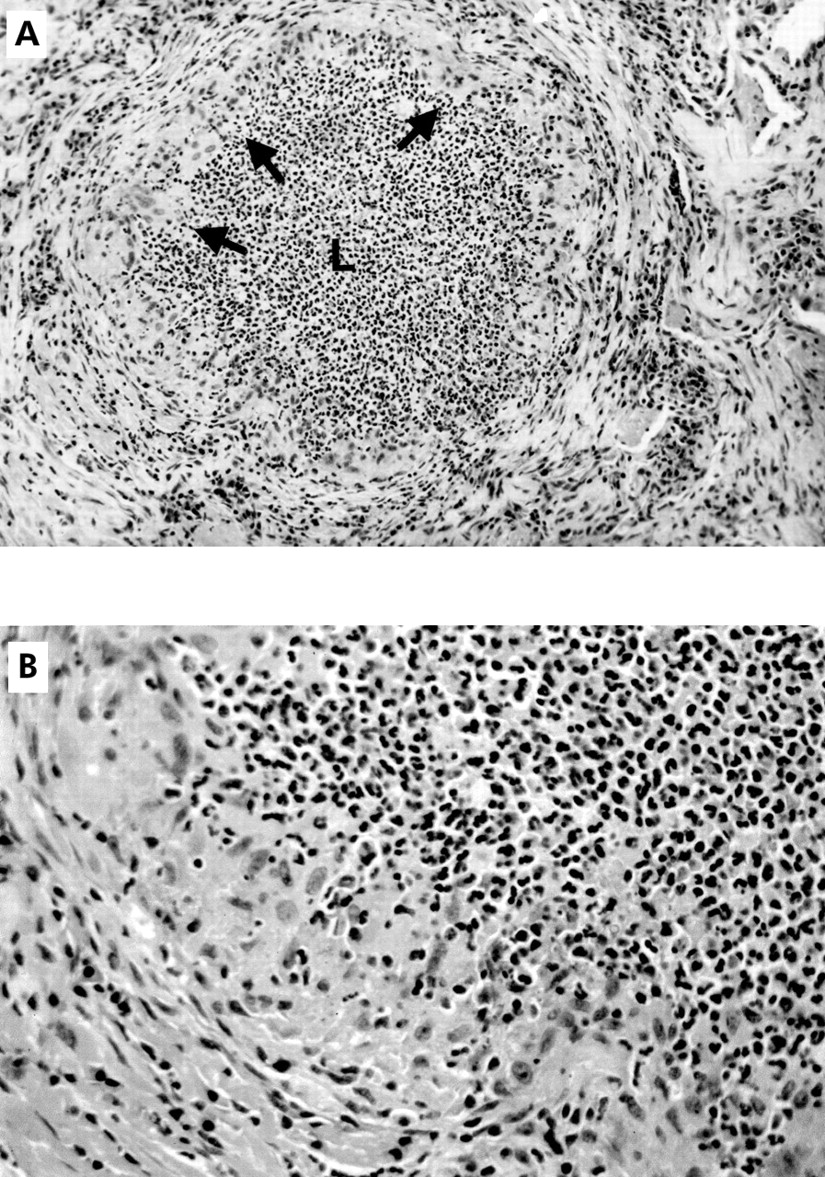
{kind=link}
(A) Lung biopsy specimen showing a large bronchiole. The lumen (L) is filled with a polymorphonuclear leucocytic exudate and the mucosa is replaced by epithelioid histiocytes (arrows). Stain: haematoxylin and eosin; original magnification ×160. (B) Section of a large bronchiole from (A) at a higher magnification showing the polymorphonuclear leucocytes. Stain: haematoxylin and eosin; original magnification ×400.
She was started on intravenous methylprednisolone 20 mg every 8 hours and oral itraconazole 400 mg daily with rapid resolution of the fever. Follow up over the subsequent 3 months revealed marked symptomatic and objective improvement (TLC 92%, FVC 89%, FEV1 95%, Tlco 64% predicted). The chest radiograph showed resolution of the interstitial pattern and the IgE level declined to 2500 IU/ml. She continues to do well 3 years later.
Her brother’s necroscopic slides from 23 years earlier were reviewed and it was found that he had chronic granulomatous disease (CGD). Accordingly, the patient’s blood was sent for studies of neutrophil function. The dihydrorhodamine flow cytometry assay revealed that only 10% of her neutrophils had a normal oxidative burst in response to phorbol myristate acetate. Immunoanalysis using specific antibodies to four structural components of NADPH oxidase showed that only 10% of her neutrophils expressed detectable levels of gp91phox and p22phox. The other two subunits, p47phox and p67phox, were unaffected. These findings are compatible with an X-linked CGD carrier state with skewed X chromosome inactivation. The mutation responsible for this defect in the CYBB gene which encodes gp91phox was localised to exon 7 of the X chromosome.
METHODS
Histological preparation
Lung sections were immersion fixed in 10% neutral buffered formalin overnight. Tissues were processed and embedded in paraffin and 4 μm thick sections were stained with haematoxylin and eosin (H&E), elastic trichrome, silver methenamine, and Ziehl-Neelsen stain.
Neutrophil oxidative burst and NADPH subunit analysis
A flow cytometric assay of neutrophil oxidant production was done using dihydrorhodamine as previously described except that dichlorofluorescein was replaced by dihydrorhodamine (Molecular Probes, Eugene, OR, USA) as the detection agent.3,4 Expression of individual protein subunits of the NADPH oxidase complex was identified in neutrophils from the patient and normal control subjects by immunoassay using flow cytometry and monoclonal antibodies specific for each of the four subunit proteins (J Rae, unpublished data).
CYBB gene sequencing
DNA was isolated from whole blood collected into EDTA using the Puregene DNA isolation kit (Gentra Systems, Inc., Minneapolis, MN, USA). All 13 exons and the 5′ flanking region of the gene for NADPH oxidase were analysed by single strand conformation polymorphism (SSCP) analysis using single polymerase chain reactions (PCRs) with 18–27 bp oligonucleotide primers (sequences available upon request) and 500 ng genomic DNA. The resultant products were analysed on Novex 20% TBE gels using a ThermoFlow unit (Novex, San Diego, CA, USA) and stained with SYBR Green II RNA gel stain (Molecular Probes, Eugene, OR, USA). SSCP identified exon 7 as likely to contain a heterozygous nucleotide change. Amplification of the exon by PCR was performed as described by Rae et al.5 PCR amplified fragments were purified using a QIAquick PCR purification kit (Qiagen, Valencia, CA, USA) and analysed by direct sequencing in both directions using the ABI Prism BigDye terminator cycle sequencing ready reaction kit (PE Applied Biosystems, Foster City, CA, USA) and an ABI Prism 310 genetic analyser.
DISCUSSION
We report a non-asthmatic woman with biopsy proven BCG and A fumigatus colonisation in association with CGD. There was no evidence of invasive A fumigatus infection on the lung biopsy specimen. To our knowledge, this is only the second report in the literature of a patient with impaired neutrophil oxidase activity who developed the clinical syndrome of BCG. Barrot et al recently described a 38 year old non-asthmatic woman who developed BCG and A fumigatus colonisation.6 A family history of CGD resulted in the diagnosis of a rare variant of autosomal recessive disease (deficiency of p67phox).
CGD is caused by a number of mutations in the components of NADPH oxidase leading to defects in superoxide generation.7 This results in neutrophils with a defective respiratory burst during phagocytosis and defective microbial killing. The mutations may affect any of the four structural components of NADPH oxidase: membrane bound glycoprotein gp91phox and p22phox or the cytoplasmic components p47phox and p67phox. These mutations are inherited in an X-linked pattern in two thirds of patients and by autosomal recessive inheritance in the remaining one third. Defects in gp91phox, the most frequent abnormality, result from mutations in the CYBB gene located on the X chromosome. CGD manifests clinically as recurrent suppurative infections with catalase positive bacteria such as Staphylococcus and fungi, especially A fumigatus.
Despite its high mortality, one quarter of all the living patients in the US CGD National Registry were 20 years of age or older.8 Liese et al reported a series of 11 patients in whom the diagnosis was made later in life (median age at diagnosis 22 years, range 13–43).9 The median age of the first clinical manifestation of the disease was 3.6 years (range 0.1–22.5). The initial manifestations included pneumonia (especially with A fumigatus), recurrent lymphadenitis, liver or cutaneous abscesses, and sepsis.
Our patient is a carrier of the X-linked form of the disease and the severe phenotype of CGD in her brother is also consistent with the X-linked mode of inheritance. Heterozygous females have bactericidal capacity in between affected patients and normal subjects. Increased phenotypic expression of CGD in this patient may be due to skewed lyonisation (inactivation) of the normal X chromosome. If fewer than 10% of the neutrophils function normally, then there is an increased risk of infections, albeit usually mild.10
The patient’s previous illnesses were probably manifestations of her X-linked CGD carrier state. Discoid lupus erythematosus-like lesions have been reported in female carriers of X-linked CGD.11 It is possible that her “ulcerative colitis” was a bowel manifestation of her impaired neutrophil oxidase, which can produce a granulomatous colitis that mimics inflammatory bowel disease.12
In patients with BCG and a history of asthma, the presence of Aspergillus in the airways has been observed in many of the biopsy specimens. It has been postulated that the disease in this group represents a hyperimmune reaction to colonising fungi similar to allergic bronchopulmonary aspergillosis (ABPA).2,13 In non-asthmatic patients with BCG, an aetiological agent is generally not found. Our patient did not fulfil the criteria for ABPA. In particular, she did not have a history of asthma and there was no bronchiectasis on the high resolution CT scan. There was no evidence of tissue invasion, as might be seen with invasive aspergillosis in the setting of more profound neutrophil dysfunction. This combination of findings strongly suggests a hypersensitivity reaction to A fumigatus as the cause for her BCG.
BCG has usually been treated with steroids alone, although spontaneous remissions have been reported.13 In our patient an antifungal agent (itraconazole) was used in addition to corticosteroids for two reasons. Firstly, recent reports have suggested that antifungal agents such as itraconazole are beneficial in the treatment of ABPA.14,15 Secondly, we were concerned about the risk of invasive A fumigatus infection in this patient given her immunodeficiency state in combination with glucocorticoid therapy. BCG has previously been described in three immunocompromised transplant recipients who progressed to develop an invasive Aspergillus infection.16 The other reported case of BCG with CGD did not respond to treatment until antimycotics were added to the corticosteroids.6,16 Conrad et al described a 32 year old patient with CGD who developed a fatal diffuse granulomatous pneumonia after shovelling wood chips.17 Pending further studies, we recommend the consideration of chronic antifungal prophylaxis for patients with CGD if there is evidence of colonisation with Aspergillus species.
In summary, our patient is an X linked CGD carrier with skewed inactivation of the normal X chromosome causing impaired neutrophil function. We speculate that exposure to A fumigatus in this setting of ineffective immune response resulted in persistent colonisation and the subsequent development of a hypersensitivity reaction that manifest as BCG. The co-existence of these two very rare diseases is unlikely to be coincidental and it raises the question of whether there is a broader link between abnormal phagocyte function and the development of hypersensitivity reactions.
Acknowledgments
This work was supported in part by NIH grants CA68276 and AI24838.
REFERENCES
Footnotes
-
This work was supported in part by a CIHR grant MT–10994 to G Downey and NIH grants CA68276 and AI24838 to P G Heyworth and J Rae.