Article Text

Abstract
Background: Because injury to the alveolar epithelial barrier is a characteristic feature of acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS), plasma surfactant protein levels may have prognostic value. To test this hypothesis plasma surfactant proteins A and D (SP-A and SP-D) levels were measured in patients with ALI or ARDS enrolled in the NHLBI sponsored multicentre ARDS Network randomised controlled trial of a 6 ml/kg v 12 ml/kg tidal volume strategy.
Methods: Data from 565 participants in the clinical trial were used. Plasma levels of SP-A and SP-D were measured at baseline and on day 3 after the start of the mechanical ventilation protocol. The longitudinal impact of baseline plasma surfactant protein levels on clinical outcomes was examined by multivariate analysis, controlling for mechanical ventilation group, APACHE III score, and other clinical covariates. The effect of 6 ml/kg tidal volume ventilation on plasma SP-A and SP-D levels was evaluated using analysis of covariance.
Results: Baseline plasma SP-A levels were not related to any clinical outcome. In contrast, higher baseline plasma SP-D levels were associated with a greater risk of death (OR 1.21 per 100 ng/ml increment; 95% CI 1.08 to 1.35), fewer ventilator-free days (mean decrease −0.88 days; p=0.001), and fewer organ failure-free days (mean decrease −1.06 days; p<0.0001). The 6 ml/kg tidal volume strategy had no effect on the rise in plasma SP-A levels (p=0.91) but attenuated the rise in plasma SP-D levels (p=0.0006).
Conclusions: Early in the course of ALI/ARDS an increased level of plasma SP-D is associated with a worse clinical outcome. The 6 ml/kg tidal volume strategy attenuated the rise of SP-D early in the clinical course. Taken together, these observations indicate that plasma SP-D, a product of alveolar type II cells, is a valuable biomarker in ALI/ARDS.
- acute respiratory distress syndrome
- acute lung injury
- plasma surfactant associated proteins
Statistics from Altmetric.com
Despite improvements in intensive care, acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS) remain important causes of death among critically ill adults.1–3 Until recently there have been no specific treatments to reduce the mortality from ALI or ARDS. In a multicentre randomised controlled trial, the National Heart, Lung, and Blood Institute’s ARDS Network showed that a mechanical ventilation strategy using 6 ml/kg of predicted body weight reduced mortality in ALI/ARDS.4 Elucidating the mechanisms that mediate clinical outcomes, including the beneficial effects of this mechanical ventilation strategy, could provide insight into the pathophysiology of ALI/ARDS and facilitate further treatment advances.
Examination of circulating surfactant proteins A and D (SP-A and SP-D) may illuminate the impact of the 6 ml/kg ventilation strategy on clinical outcomes. These proteins are normal constituents of surfactant, which is a complex mixture of phospholipid and associated proteins. Secreted primarily by alveolar epithelial type II pneumocytes, plasma surfactant protein A and D levels appear to increase early in the clinical course of ARDS.5–7 Recent experimental models of ALI also indicate that plasma surfactant proteins are increased8 and that mechanical ventilation can modulate surfactant protein expression.9 Because the levels of these proteins progressively increase early in ALI/ARDS, they may reflect pulmonary epithelial injury and consequent increased permeability. As a consequence, analysis of SP-A and SP-D in the plasma of patients with early ALI/ARDS could provide valuable insight into the biological impact of the 6 ml/kg ventilation strategy.
To examine the longitudinal association between surfactant protein levels and the risk of death and other adverse health outcomes, we evaluated plasma samples from participants in the ARDS Network randomised controlled trial of a 6 ml/kg v 12 ml/kg tidal volume. We also evaluated the early effects of the 6 ml/kg ventilation strategy on surfactant protein levels early in the course of treatment. In particular, we hypothesised that this lower tidal volume strategy would attenuate the release of surfactant proteins into the vascular compartment.
METHODS
Study design/patient selection
Data from 565 of 861 patients participating in the National Heart, Lung, and Blood Institute ARDS Network multicentre randomised controlled trial of 6 ml/kg v 12 ml/kg tidal volume were used.4 The protocol was approved by the institutional review board at each hospital; informed consent was obtained as previously described.4 The results of the main trial have been previously reported.4 Using a factorial design, this trial was conducted simultaneously with two other clinical trials comparing ketoconazole with placebo and lisofylline with placebo. As previously published, neither medication had any clinical benefit in terms of mortality or other clinical end points.10,11 There was also no interaction between treatment with either medication and ventilator strategy assignment.4 In the present study, plasma samples were available for surfactant protein analysis from 565 of 627 patients (90%) enrolled after completion of the ketoconazole trial. There were no statistical differences between subjects with and without plasma surfactant measurements, including mechanical ventilation strategy assignment, APACHE III scores, and Pao2/Fio2 ratios (p>0.30 in all cases).
Clinical trial procedures
The study methods have been previously reported in detail, including inclusion and exclusion criteria and mechanical ventilation procedures.4,12,13 Briefly, intubated mechanically ventilated patients were eligible if there was acute onset of: (1) Pao2/Fio2 ⩽300 (altitude adjusted in Denver and Salt Lake City), (2) a chest radiograph showing bilateral pulmonary infiltrates consistent with oedema, and (3) no clinical evidence of left atrial hypertension. If measured, pulmonary capillary wedge pressure was required to be ⩽18 mm Hg. Participating subjects were randomly assigned to either the 6 or 12 ml/kg tidal volume study group. Volume assist control mode was used according to a previously described protocol.4,12,13
Surfactant protein measurement
Plasma surfactant protein measurements were made by investigators who were blinded to the ventilator group, clinical status, and health outcomes of all trial participants. Both SP-A and SP-D were measured in baseline plasma samples before implementing the ventilator protocol and at day 3.
SP-A was measured using a previously described sandwich enzyme-linked immunosorbent assay (ELISA) technique which used two different monoclonal antibodies to human SP-A (designated 9.5 and 22.7).5,14 SP-A isolated from the bronchoalveolar lavage (BAL) fluid of patients with alveolar proteinosis was used as the standard.
To measure SP-A, immunoassay plates (96 well EIA; Costar, Cambridge, MA, USA) were coated overnight at 4°C with capture antibody (mAb 9.5) diluted in 0.1 M NaHCO3 (pH 8.3) to a final concentration of 5 µg/ml. The plates then were washed twice with phosphate buffered saline (PBS)/0.2% bovine plasma albumin (BSA)/0.05% Tween-20, and blocked using the same solution for 1 h at room temperature. The detergent Triton-X (1.0% final concentration) was added to each plasma sample and standard, and the samples and standards were then probe sonicated for 5 s on ice. Samples and standards were immediately diluted in PBS/0.2% BSA/0.05% Tween-20/1% Triton X and added to the wells. The plates were incubated at 37°C for 2 h, washed three times, and the biotinylated detecting antibody (mAb 22.7) (1:250 dilution) was added to each well. After incubation for 2 h at 37°C, the wells were washed three times, incubated with HRP/strepavidin (Zymed Laboratories, San Francisco, CA, USA) for 30 min, and developed with TMB/H2O2 (Kirkegaard & Perry Laboratories, Gaithersburg, MD). The optical density in each well was read at 490 nm (OD 490) using a microtitre plate spectrophotometer (Dynatech Laboratories, Chantilly, VA). The standard curve is linear from 1 to 100 ng/ml SP-A. All assays were performed in duplicate dilutions.
Human SP-D was measured with ELISA kits (Nagae Corporation, Tokyo, Japan). Using previously described methods,5,14 the ELISA assay used recombinant human SP-D (rhSPD) as the standard and two monoclonal antibodies against human SP-D, designated 6B2 and 7C6, which were raised against human SP-D purified from BAL fluids of patients with alveolar proteinosis.15 The rhSP-D standard or samples were diluted 1:10 to 1:200, added to wells coated with antibody 7C6, and incubated overnight at 4°C. After washing, the HRP-conjugated 6B2 antibody was added and the plates were incubated at room temperature for 2 h. The plates were developed with TMB/H2O2 and the OD 450 was measured in each well. All assays were performed in duplicate.
Other clinical variables
Clinical data were reviewed and recorded at baseline and on days 1, 2, 3, 4, 7, 14, 21, and 28 between 06.00 and 10.00 hours. These data included laboratory data, ventilator parameters, Pao2/Fio2 ratio, and vasopressor use. Baseline APACHE III scores were calculated.16 For each patient the clinical coordinator and physician investigator assessed the predominant clinical risk factor for ALI/ARDS within 36 h of onset. The clinical risk factor was ascertained prospectively, before randomisation to ventilator treatment group. Pneumonia and aspiration were classified as direct pulmonary risk factors for ALI/ARDS, as opposed to non-pulmonary risk factors (sepsis, trauma, and other).13
Study outcome variables
To evaluate health outcomes, patients were followed to day 180 or until discharge home with unassisted breathing. At 180 days follow up was 99% complete. Patients alive in other health care facilities at 180 days were considered to have survived. In the present study the primary outcome was mortality before discharge home with unassisted breathing. This outcome, which was also a primary study outcome in the main clinical trial report,4 was chosen to reflect death directly attributable to ALI/ARDS.
Secondary study outcomes included the number of days without non-pulmonary organ failure.17 As in the main clinical trial,4 the number of organ failure-free days was calculated by subtracting the number of days with organ failure from the lesser of 28 days or the number of days until death. Circulatory failure was defined as any systolic blood pressure ⩽90 mm Hg or any vasopressor treatment; coagulation failure as any platelet count ⩽80000/μl; hepatic failure as any bilirubin concentration ⩾2 mg/dl (34.2 μmol/l); renal failure as any plasma creatinine concentration ⩾2 mg/dl (177 μmol/l); and neurological failure as a Glasgow coma score of ⩽12.
Other secondary outcomes included the number of ventilator-free days, which was defined as the number of days of unassisted breathing from day 1 to 28 if unassisted breathing continued for ⩾48 consecutive hours (this was considered a primary study outcome in the original clinical trial report).4 For patients who died, ventilator-free days were calculated as the number of days alive minus the duration of mechanical ventilation. A difference in ventilator-free days could reflect a difference in mortality, in the duration of mechanical ventilation, or both.
Statistical analysis
Data analysis was conducted using SAS 8.2 (SAS Institute, Cary, NC). Because plasma surfactant protein levels were not normally distributed, bivariate comparisons were conducted using the Wilcoxon and Kruskal-Wallis tests. Linear and logistic regression analyses were used for multivariate analysis. Standard regression model diagnostics were performed.
To assess the first analytical aim we examined the longitudinal impact of baseline plasma surfactant protein levels on patient outcomes. Logistic regression analysis was used to evaluate the association between baseline SP-A and SP-D levels and the risk of death, controlling for the ventilator group (6 v 12 ml/kg). In regression analysis, plasma surfactant protein levels were treated as continuous predictor variables. To facilitate presentation of results, we calculated odds ratios for each 100 ng/ml increment of SP-A or SP-D, which we defined as a meaningful change a priori. In a forced entry model we then controlled for other covariates that reflect acute illness severity, including baseline APACHE III scores, renal function (creatinine), and platelet count. We also controlled for Pao2/Fio2 ratio, a marker for the severity of acute lung injury. Each of these covariates was associated with mortality among the cohort of ALI/ARDS patients (p<0.05 in all cases). Because lisofylline and ketoconazole are not related to clinical outcomes,10,11 they were not considered as confounding variables. To examine the longitudinal impact of baseline surfactant proteins on ventilator-free days and organ failure-free days, we used multivariate linear regression in analogous fashion. Results were expressed as the average change in outcome per 100 unit increment in SP-A or SP-D level.
In addition to the analysis evaluating surfactant proteins as continuous predictor variables, we divided plasma SP-A and SP-D levels into quartiles and examined the impact of surfactant protein quartile on clinical outcomes. An advantage of this analysis is that it does not assume a linear relation between surfactant protein levels and patient outcomes. The Mantel-Haenszel χ2 test was then used to test a linear exposure-response trend between plasma surfactant protein levels and clinical outcomes.
To evaluate the second analytical aim we studied the prospective impact of the 6 ml/kg tidal volume strategy on the change in surfactant protein levels over time. Analysis of covariance (ANCOVA) was used to examine the effect of mechanical ventilator treatment group on surfactant protein levels measured at day 3, controlling for baseline level. To address the non-normal distribution of surfactant protein levels, we performed ANCOVA on rank transformed SP-A and SP-D levels. Because some study subjects died or were extubated between day 0 (baseline) and day 3, not all study subjects had surfactant protein measurements at day 3. To test the impact on study results we imputed missing surfactant protein data at day 3 by carrying forward the previous value. There was no appreciable impact on the results and these data are not presented.
Because the clinical risk factor for ALI/ARDS influences the likelihood of death,13 we also studied the impact of clinical risk factor on the relationship between baseline surfactant protein levels and the risk of death. To evaluate for statistical interaction between surfactant protein levels and death, we used the likelihood ratio test to compare logistic regression models, with and without interaction terms, between surfactant protein levels and clinical risk group. Because the power to detect statistical interactions is generally low, we used a cut off α (two tailed) of 0.10 to indicate a statistically significant interaction.18
RESULTS
Baseline characteristics
Table 1shows the baseline demographic and clinical characteristics of 565 patients with ALI or ARDS in whom plasma surfactant protein levels were measured. At baseline (ventilator protocol initiation) the median plasma SP-A level was 30 ng/ml (range 0–495 ng/ml; 25–75th interquartile range (IQR) 14–64 ng/ml) and the median SP-D level was 82 ng/mg (range 3–1061 ng/ml; 25–75th IQR 38–166 ng/ml).
Baseline characteristics of 565 patients with acute lung injury/acute respiratory distress syndrome
Baseline plasma surfactant protein levels and patient outcomes
At baseline, plasma SP-A levels were similar among patients who went on to survive or die (p=0.69; table 2). In contrast, baseline plasma SP-D levels were higher in patients who subsequently died (median 101 ng/ml) than in survivors (median 73 ng/ml, p=0.013; table 2).
Baseline surfactant protein levels and mortality among patients with ALI/ARDS
Baseline SP-A levels were not associated with any clinical outcome, after controlling for tidal volume group (table 3). In contrast, higher baseline SP-D levels were associated with a greater risk of death among patients with ALI/ARDS (OR per 100 ng/ml increment 1.16; 95% CI 1.05 to 1.29). Higher baseline SP-D levels were also associated with fewer ventilator-free days (mean decrease −0.97 days; 95% CI −1.49 to −0.44) and fewer organ failure-free days (−0.94 days; 95% CI −1.48 to −0.40). After controlling for the other clinical covariates, higher baseline SP-D levels were related to a greater risk of death (OR 1.21; 95% CI 1.08 to 1.35) and fewer ventilator-free and organ failure-free days.
Baseline surfactant protein level and outcomes among patients with ALI/ARDS
To further examine the impact of SP-D, the analysis was repeated using baseline SP-D quartiles, controlling for ventilator group. Compared with the lowest quartile, the risk of death increased progressively for the second, third, and fourth quartiles (OR 0.96, 95% CI 0.57 to 1.60; OR 1.43, 95% CI 0.87 to 2.37; and OR 1.97, 95% CI 1.21 to 3.22). The Mantel-Haenszel χ2 test was consistent with a linear exposure response trend (p=0.002).
As baseline SP-D quartile increased, mean ventilator-free days also decreased progressively for each successive quartile (compared with the lowest quartile): −0.73 days (95% CI −2.09 to 2.83), −2.16 days (95% CI −4.66 to 0.34), and −4.34 days (95% CI −6.81 to −1.87). Organ failure-free days also decreased as the baseline SP-D level increased: 0.59 days (95% CI −1.96 to 3.14), −2.63 days (95% CI −5.21 to −0.043), and −3.53 days (95% CI −6.09 to −0.96). In both cases there was evidence of linear exposure-response trend for SPD (p<0.001 for both). For each clinical outcome the risk was greatest for the highest SP-D quartile.
Tidal volume strategy and surfactant protein levels
Among the entire group of 565 patients with ALI/ARDS, SP-A levels increased during the first three study days (table 4). However, there was no apparent difference in the quantitative increase between the 6 ml/kg and 12 ml/kg tidal volume groups (table 4). Compared with the 12 ml/kg tidal volume strategy, the 6 ml/kg tidal volume strategy did not appreciably affect the change in plasma SP-A levels (0.2 ng/ml; 95% CI −10 to 10).
6 ml/kg v 12 ml/kg tidal volume strategies and surfactant protein levels
SP-D levels also increased during the first three days after study initiation (p<0.0001; table 4, fig 1). Compared with the 12 ml/kg tidal volume strategy, the 6 ml/kg tidal volume strategy attenuated this early rise in SP-D (p=0.0006; table 4, fig 1). On average, plasma SP-D levels rose less in the 6 ml/kg tidal volume group than in the 12 ml/kg tidal volume group (−66 ng/ml; 95% CI −36 to −97).

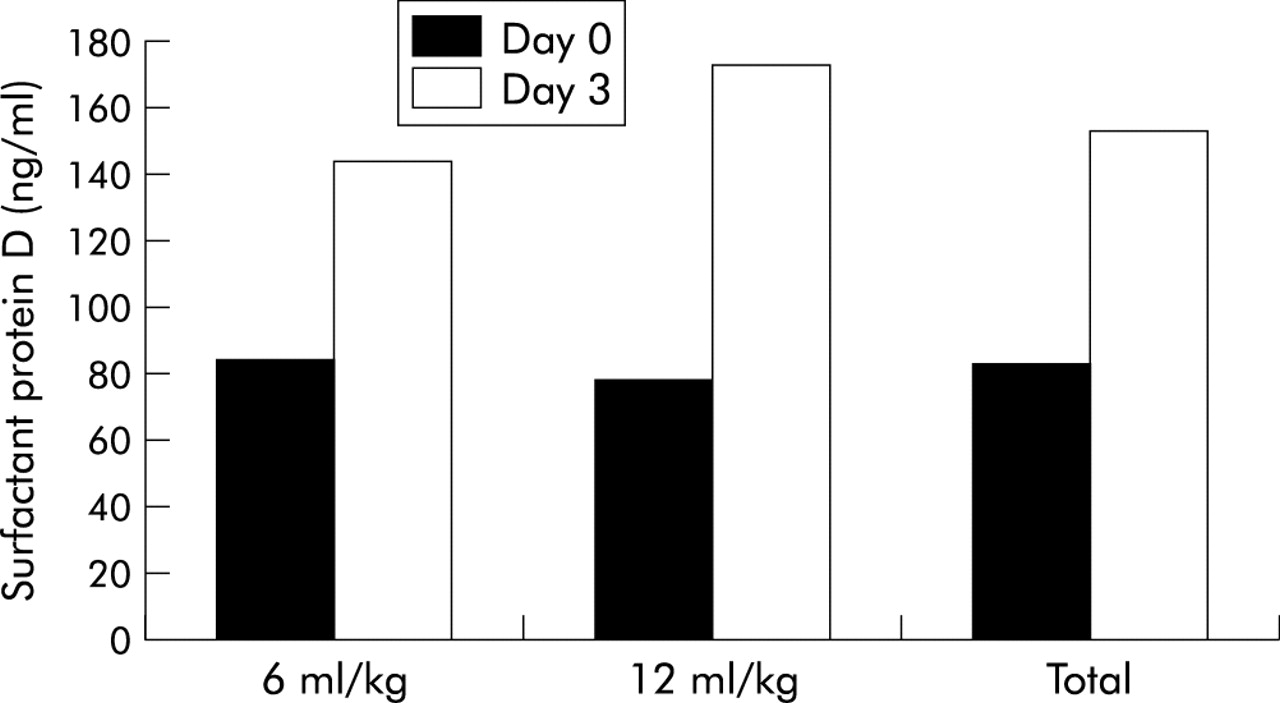{kind=link}
Impact of low tidal volume versus traditional tidal volume strategy on plasma SP-D levels in patients with ALI/ARDS. The figure shows plasma SP-D levels at baseline (day 0), before institution of mechanical ventilation protocol, and on day 3. Compared with the traditional tidal volume strategy, the low tidal volume strategy attenuated the early rise in SP-D (p=0.0006). See table 4for median values (and interquartile ranges) of plasma SP-D levels in each ventilator strategy and at each time point.
Clinical risk group for ALI/ARDS and surfactant protein levels
Baseline surfactant protein levels varied by the clinical risk factor for ALI/ARDS (table 5). SP-A levels were highest among patients with pneumonia (median 39 ng/ml; 25–75th IQR 20–70 ng/ml) and lowest in those with trauma (median 24 ng/ml; IQR 11–42 ng/ml, p=0.003). Similarly, SP-D levels were highest in the patients with pneumonia (median 113 ng/ml; IQR 51–280 ng/ml) and lowest in those with trauma (median 54 ng/ml; IQR 36–89 ng/ml, p<0.0001).
Baseline surfactant protein level by clinical risk factor for ALI/ARDS
To further investigate the impact of baseline SP-D levels we examined whether the relationship between SP-D levels and a greater risk of death varied by the clinical risk factor for ALI/ARDS. There was evidence of statistical interaction between baseline SP-D level and clinical risk group (p=0.054). The association between baseline SP-D levels and risk of death was strongest for sepsis (OR 1.43; 95% CI 1.14 to 1.80) and pneumonia (OR 1.13; 95% CI 1.005 to 1.28). There was no clear statistical relation between SP-D levels and death in the aspiration (OR 1.08; 95% CI 0.85 to 1.36) or other groups (OR 1.04; 95% CI 0.79 to 1.36). Among patients with trauma, higher baseline SP-D levels were related to a lower risk of death, although the confidence interval was wide and did not exclude an association (OR 0.63; 95% CI 0.28 to 1.40).
DISCUSSION
In a randomised controlled trial of two mechanical ventilation strategies for the treatment of ALI/ARDS, higher baseline levels of plasma SP-D were associated with worse clinical outcomes, including a greater risk of death, fewer ventilator-free days, and fewer organ failure-free days. Furthermore, the 6 ml/kg tidal volume strategy attenuated the rise of plasma SP-D levels early in the course of treatment. In contrast, plasma SP-A levels were not related to clinical outcomes in early ALI/ARDS.
These results show that SP-D may be a valuable plasma biomarker of lung injury in patients with ALI/ARDS. Recent experimental work in rats indicated that increased plasma SP-D levels could reflect type II cell hyperplasia, acute lung injury, or both.8 Intrabronchial instillation of keratinocyte growth factor, which produces type II cell hyperplasia, resulted in increased plasma SP-D levels.8 In a hydrochloric acid model in which acute lung injury develops before type II cell proliferation, plasma SP-D levels were also raised.8 Our finding that plasma SP-D levels are increased early in ALI/ARDS is most consistent with increased alveolar epithelial permeability because significant type II cell hyperplasia would not be expected this early in the clinical course of ALI/ARDS. SP-D may be a more sensitive indicator of alveolar epithelial permeability than SP-A because of its more hydrophilic nature, allowing it to enter the vascular compartment more readily.
Previous investigators have shown that serum SP-A and SP-D levels are increased early in the course of ALI/ARDS.5–7 Moreover, plasma SP-A levels predict the development of ARDS in patients at risk for the syndrome.14 The relationship between plasma surfactant levels and clinical outcomes in ARDS, however, has been less clear. Using data from 36 ARDS patients, Greene and colleagues found no clear evidence that plasma SP-A or SP-D levels predicted mortality.5 Another small single centre study of 38 patients also found no clear impact of these plasma surfactant proteins on mortality in patients with ARDS.19 In contrast, some clinical studies have shown that higher plasma SP-A6 and SP-D levels5 are related to more severe lung injury. These studies, however, were based on small numbers of patients and did not have adequate statistical power to examine the impact of surfactant proteins on mortality and other important clinical outcomes.
A previous randomised controlled trial of 44 patients found that a lung protective ventilation strategy attenuated the levels of proinflammatory cytokines in plasma and BAL fluid.20 Based on this observation, Ranieri and colleagues hypothesised that a lung protective ventilation strategy designed to limit plateau pressure may reduce disruption of the pulmonary epithelial-endothelial barrier. Their data, however, did not directly evaluate the alveolar barrier. Our finding that higher plasma SP-D levels are related to adverse clinical outcomes supports the theory that injury to the alveolar epithelial barrier is fundamental to the pathogenesis of ALI/ARDS. Because the 6 ml/kg tidal volume strategy attenuated the rise of plasma SP-D levels, our findings also indicate that the reduction of alveolar epithelial injury may mediate, at least in part, the benefit of this tidal volume strategy. Recent experimental work further supports the salutary impact of lung protective ventilation on the alveolar epithelium.9 Alternative explanations could include an effect of mechanical ventilation strategy on SP-D synthesis or on rate of clearance of SP-D from the circulation.
One limitation of the present study is that, for logistic reasons, we examined a subset of clinical trial participants. Because we evaluated 90% of patients participating in the latter two thirds of the randomised controlled trial, we do not anticipate that this study method introduced selection bias. Moreover, subjects with plasma surfactant measurements were similar to the other trial participants in terms of mechanical ventilation strategy assignment and acute illness severity (APACHE III scores). The observed relationship between baseline surfactant proteins and subsequent clinical outcomes could also be affected by confounding factors such as the severity of illness. Arguing against this possibility, controlling for APACHE III score and other indicators of acute illness, severity did not alter the conclusions. In addition, some study subjects who had baseline surfactant protein measurements had no day 3 measurements due to death and liberation from mechanical ventilation. This did not appear to introduce bias, as a repeated analysis carrying forward the previous surfactant protein value did not appreciably impact the results (data not shown). Finally, the findings derived from this randomised controlled trial may not be generalisable to the overall population of patients with ALI/ARDS. However, the clinical features of trial participants are similar to those of previous cohort studies of patients with ALI/ARDS, supporting the generality of the present results. Although plasma SP-D appears to be a promising biomarker in ALI/ARDS, its value should be confirmed in another independent population of ALI/ARDS patients.1,2,21–28
Higher baseline plasma SP-D levels are associated with adverse clinical outcomes in ALI/ARDS. The 6 ml/kg tidal volume strategy, which reduces mortality in these conditions, attenuates the rise of plasma SP-D levels early in the course of treatment. Taken together, these observations suggest that SP-D may serve as a valuable pulmonary-specific biomarker of injury to the alveolar epithelium. Plasma SP-D levels may therefore be useful in future clinical trials of treatments for ALI or ARDS.
APPENDIX: National Institutes of Health National Heart, Lung, and Blood Institute ARDS Network
Network participants: Cleveland Clinic Foundation—H P Wiedemann*, A C Arroliga, C J Fisher Jr, J J Komara Jr, P Periz-Trepichio; Denver Health Medical Center— P E Parsons; Denver VA Medical Center—C Welsh; Duke University Medical Center—W J Fulkerson Jr,* N MacIntyre, L Mallatratt, M Sebastian, J Davies, E Van Dyne, J Govert; Johns Hopkins Bayview Medical Center—J Sevransky, S Murray; Johns Hopkins Hospital—R G Brower, D Thompson, J Sevransky, S Murray; LDS Hospital—A H Morris*, T Clemmer, R Davis, J Orme Jr, L Weaver, C Grissom, F Thomas, M Gleich (posthumous); McKay-Dee Hospital—C Lawton, J D’Hulst; Metro Health Medical Center of Cleveland—J R. Peerless, C Smith; San Francisco General Hospital Medical Center—R Kallet, J M. Luce; Thomas Jefferson University Hospital—J Gottlieb, P Park, A Girod, L Yannarell; University of California, San Francisco—M A Matthay,* M D Eisner, J Luce, B Daniel; University of Colorado Health Sciences Center—E Abraham,* F Piedalue, R Jagusch, P Miller, R McIntyre, K E Greene; University of Maryland—H J Silverman,* C Shanholtz, W Corral; University of Michigan—G B Toews, D Arnoldi, R H Bartlett, R Dechert, C Watts; University of Pennsylvania—P N Lanken,* H Anderson, B Finkel, William Hanson; University of Utah Hospital—R Barton, M Mone; University of Washington/Harborview Medical Center—L D Hudson,* G Carter, C Lee Cooper, A Hiemstra, R V Maier, K P Steinberg; Utah Valley Regional Medical Center—T Hill, P Thaut; Vanderbilt University—A P Wheeler,* G Bernard,* B Christman, S Bozeman, L Collins, L B Ware.
Clinical Coordinating Center: Massachusetts General Hospital, Harvard Medical School—D A Schoenfeld,* B T Thompson, M Ancukiewicz, D Hayden, F Molay, N Ringwood, G Wenzlow, A S Kazeroonin.
NHLBI Staff: D B Gail, A Harabin,* P Lew, M Waclawiw.
*Steering Committee: G R Bernard, Chair, Principal Investigator from each centre as indicated by an asterisk above.
Data and Safety Monitoring Board: R G Spragg, Chair, J Boyett, J Kelley, K Leeper, M Gray Secundy, A Slutsky.
Protocol Review Committee: J G N Garcia, Chair, S S Emerson, S K Pingleton, M D Shasby, W J Sibbald.
Acknowledgments
Support was provided by contracts (NO1-HR 46054, 46055, 46056, 46057, 46058, 46059, 46060, 46061, 46062, 46063, and 46064) with the National Heart, Lung, and Blood Institute. In addition, Dr Eisner was supported by K23 HL04201 and Dr Matthay by R01 HL51856, from the National Heart, Lung, and Blood Institute. Dr Greene was supported by HL-03724 and the Cystic Fibrosis Foundation.
REFERENCES
Linked Articles
- Airwaves