Article Text

Abstract
Background: Inhaled tumour necrosis factor alpha (TNFα ) has previously been shown to induce airway neutrophilia and increased airway reactivity in normal subjects. It was hypothesised that a similar challenge would increase airway reactivity in those with mild asthma, but that the inflammatory profile may differ.
Methods: Ten mild asthmatic subjects were recruited on the basis of clinical asthma and either a sensitivity to methacholine within the range defined for asthma or a 20% improvement in forced expiratory volume (FEV1) after 200 μg salbutamol. Subjects inhaled either vehicle control or 60 ng recombinant human (rh)TNFα and were studied at baseline, 6, 24, and 48 hours later. Variables included spirometric parameters, methacholine provocative concentration causing a 20% fall in FEV1 (PC20), induced sputum differential cell count, relative sputum level of mRNA of interleukins (IL)-4, IL-5, IL-9, IL-14, IL-15 and TNFα, and the exhaled gaseous markers of inflammation, nitric oxide and carbon monoxide.
Results: PC20 showed an increase in sensitivity after TNFα compared with control (p<0.01). The mean percentage of neutrophils increased at 24–48 hours (24 hour control: 1.1 (95% CI 0.4 to 2.7) v 9.2 (95% CI 3.5 to 14.9), p<0.05), and there was also a rise in eosinophils (p=0.05). Relative levels of sputum mRNA suggested a rise in expression of TNFα , IL-14, and IL-15, but no change in IL-4 and IL-5. Spirometric parameters and exhaled gases showed no significant change.
Conclusion: The increase in airway responsiveness and sputum inflammatory cell influx in response to rhTNFα indicates that TNFα may contribute to the airway inflammation that characterises asthma.
- asthma
- tumour necrosis factor α
Statistics from Altmetric.com
Many mediators have been implicated in the pathogenesis of allergic asthma. The initial events in previously sensitised allergic asthmatic subjects involve specific IgE cross linking after allergen inhalation, followed by the degranulation and activation of the IgE sensitised mast cell. This cell releases well characterised preformed mediators including histamine and proteases such as chymase and tryptase, as well as eicosanoids. More recently it has become clear that the mast cell stores cytokines in cytoplasmic granules. These cytokines include TNF alpha (TNFα) which can be upregulated by recognised growth factors for the mast cell such as stem cell factor (SCF, or c-kit ligand).1,2
It is becoming increasingly apparent that TNFα plays a role in asthma. TNFα mRNA is more frequently expressed in the airway of asthmatic subjects than normal subjects and increased release of this cytokine has been shown from the bronchoalveolar lavage (BAL) cells of asthmatic subjects.3 Also, lipopolysaccharide inhalation by mild asthmatic subjects induces TNFα secretion into BAL fluid and is associated with increased airway reactivity.4,5 In addition, after allergen challenge TNFα is found to be increased in the BAL fluid of asthmatic subjects, and their peripheral blood monocytes generate more of this cytokine.6,7 Previous work has shown that TNFα causes hyperresponsiveness in rats,8 and in man it has been found that exogenous TNFα can induce a neutrophil influx followed by increased airway reactivity in normal subjects.9 It was hypothesised that a similar inhalational challenge may induce an increase in airway reactivity in patients with mild asthma, and that the inflammatory influx may differ from the previous study group of normal subjects.
METHODS
The protocol was approved by the committee on ethical procedures in human subjects of the University of New South Wales. Informed consent was obtained from all participants in the trial.
Subjects and reagents
Ten mild asthmatic subjects (three women) were recruited to the study on the basis of clinical asthma without use of inhaled glucocorticosteroids in the last 3 months, and either a sensitivity to methacholine within the range defined for asthma or a 20% improvement in forced expiratory volume in the first second (FEV1) after salbutamol. None were current smokers or had a >1 pack year history of smoking. Subjects were assessed for atopy by symptom questionnaire and skin prick testing with common allergens including Felix domesticus, Dermatophagoides pteronyssinus, Aspergillus fumigatus, and mixed grasses (Bayer, Pymble, Sydney, Australia). Atopy was defined as seasonal conjunctivitis/hayfever and by a skin prick test >2 mm larger than the negative control. Table 1 shows the characteristics of the study subjects.
Characteristics of study subjects
Reagents were purchased from Sigma Australia unless otherwise specified.
Study design and protocol
The design was that of a double blind, placebo controlled, randomised, crossover study using within subject comparison. On the first study day subjects underwent spirometric testing followed by methacholine challenge and sputum induction, and then inhaled either nebulised recombinant human (rh) TNFα (60 ng, Bioscientific, Gymea, Australia) in 1% human albumin-phosphate buffered saline9 or vehicle control. Subjects were monitored for 6 hours and then a further sputum induction was performed. Methacholine challenge and sputum induction were repeated at 24 and 48 hours after test inhalation. After an interval of 6 weeks the alternate arm of the study was performed with the same variables being assessed. Variables studied were: spirometric parameters, methacholine responsiveness, cellular profile and cytokine mRNA of induced sputum, exhaled nitric oxide (eNO) and carbon monoxide (eCO).
Spirometry and bronchial responsiveness
At the same time of day, subjects initially performed baseline spirometric tests according to ATS recommendations using a dry wedge spirometer (Vitalograph; Buckingham, UK) to derive FEV1 and forced vital capacity (FVC).10 A standard methacholine (MCh) challenge test11 was then administered. Briefly, subjects inhaled 0.9% saline five times by slow inspiration from functional residual capacity to total lung capacity over 1 second using a Mefar dosimeter (Mefar, Brescia, Italy) and then held their breath for 6 seconds. FEV1 was measured 2 minutes later. Doubling concentrations of methacholine from 0.0625 to 128 mg/ml were inhaled until either a 20% fall in FEV1 occurred or the concentration of 128 mg/ml was reached. Results are expressed as the logarithm of the dose needed to cause a 20% fall in FEV1 (log PC20). Airway obstruction was reversed by 200 μg inhaled salbutamol .
Sputum induction
Sputum induction was performed using a method previously described.9 Briefly, subjects inhaled nebulised 3.5% saline and lavaged orally with water before voluntary coughing every 2.5 minutes until 5 ml sputum was obtained or 20 minutes had elapsed. Sputum was solubilised by incubation with 1 mmol dithiothreitol in phosphate buffered saline (PBS) made up to a total volume of 10 ml and incubated for 15 minutes at 37°C. Cells were then washed thrice in PBS, the cell pellet resuspended, and the total number of pulmonary cells was counted by toluidine blue staining using a haemocytometer. A sample of cells from each preparation was mounted on slides, fixed with methanol, and sequentially stained with eosin and methylene blue dyes. A differential cell count was performed by applying standard morphometric criteria to the first 400 identifiable pulmonary cells. Sputum preparation for cytokine analysis was performed as previously described.12 Briefly, sputum was stored in denaturing solution13 consisting of guanadinium, sarcosyl, and β-mercaptoethanol.
Cytokine mRNA analysis
Total RNA was extracted using the acid-guanadinium thiocyanoate-phenol-chloroform method.13 RNA was precipitated with isopropanol in the presence of glycogen, resuspended in denaturing solution, re-precipitated, and washed in ethanol. Adjustments were made to ensure that RNA from an equivalent number of pulmonary cells was used.
RNase protection assays were performed on sputum specimens according to the manufacturer's instructions (RiboQuant, Pharmingen, San Diego, CA, USA), and mRNA expression determined for TNFα , IL-2, IL-4, IL-5, IL-9, IL-13, IL-14, IL-15, and interferon (IFN)γ. Briefly, sample RNA was hybridised to the {α 32P}dUTP-labelled hCK-1 probe set and the single stranded RNA digested with a RNase A/T1 mixture before purification and electrophoretically size separated on a 5% polyacrylamide gel (PAGE). The dried gel was subjected to densitometry and phosphorimager analysis to evaluate the protected bands. Data were expressed as the ratio derived from cytokine/GAPDH density in each lane for each individual. Positive controls consisted of human RNA for each cytokine and yeast tRNA was a negative control; RNA quality was assessed by PAGE.
Exhaled nitric oxide
Exhaled nitric oxide (eNO) was measured online using a chemiluminescence analyser (Dasibi, Glendale, CA, USA) adapted for online measurement by the method of Kharitonov et al,14 which has been verified as uncontaminated by nasal NO15 and complies with current standards published by the ATS and ERS.16,17 Subjects exhaled using a slow vital capacity manoeuvre maintaining a positive oral pressure to exclude nasal contamination, and the mean of three peak plateau readings was taken.
Exhaled carbon monoxide levels (eCO) were also measured simultaneously (Leybold-Heraeus Binos 1, Hanau, Germany) via a side arm and the peak plateau levels were recorded. NO free gas and standard concentrations of NO and CO gases (BOC, Sydney) were used to calibrate the analysers.
Statistical analysis
Previous work using the primary end point of change in methacholine log PC20 sensitivity and a standard deviation of 0.22 suggested that a significant change would be detected with eight subjects (α=0.05, 1–β=0.8).9 Data are expressed as mean (95% confidence interval, CI) unless otherwise indicated, while log data are expressed as geometric means. Statistical analysis was performed by repeated measures analysis of variance (ANOVA) for data conforming to the Normal distribution with significant results being further analysed by post hoc Bonferroni corrected t tests for multiple comparisons. The Wilcoxon sign rank test was applied to data with a non-parametric distribution.18
RESULTS
Subjects
All of the subjects tolerated the procedures without incident, but unfortunately one subject failed to complete one arm of the study despite repeated invitations to return. The data are analysed using the remaining nine subjects.
Spirometry
There was a small fall in mean FEV1 from 3.55 l (95% CI 1.82 to 5.28) to 3.50 l (95% CI 1.7 to 5.33) 24 hours after rhTNFα (control: from 3.59 l (95% CI 1.63 to 5.5) to 3.56 l (95% CI 1.8 to 5.2)). This did not reach statistical significance. FVC did not show any significant change at any of the time points.
Methacholine responsiveness
Comparing the TNFα inhalation phase with the saline vehicle phase, there was a significant difference between the two groups. The same subjects showed an increase in sensitivity to methacholine after inhaling rhTNFα, and the difference between the two treatments reached statistical significance at 24 hours, as shown in fig 1 (mean difference 83.2%, 95% CI 33.3 to 133.1, Bonferroni corrected post hoc t test, p<0.05). Repeated methacholine challenge led to a decrease in sensitivity during the control arm, as previously observed.9,11 All subjects achieved a MCh PC20, including the two subjects with a baseline value of >16 mg/ml who required an upper doubling dilution of 128 mg/ml.

Effect of control or tumour necrosis factor alpha (TNFα) inhalation upon log PC20 expressed as percentage of the baseline value after 24 and 48 hours. Open symbols=control challenge; closed symbols=60 ng rhTNFα challenge. Mean and SE values are also shown (ANOVA, p<0.01; post hoc Bonferroni corrected t test, p<0.05 at 24 hours).
Sputum inflammatory differential cell counts
After TNFα inhalation there was a significant increase in the percentage of sputum neutrophils at 24 hours (mean difference 8.1%, 95% CI 6.3 to 9.9, post hoc Bonferroni corrected t test, p<0.05) and at 48 hours (mean difference 3.4%, 95% CI 1.8 to 5.0, Bonferroni corrected t test, p<0.02) and an increase also in eosinophils at 24 hours (mean difference 1.5%, 95% CI 1.3 to 1.7, Bonferroni corrected t test, p=0.05) compared with the control arm of the study (fig 2). The maximum increase in both of these inflammatory cells was seen at 24 hours and was beginning to subside by 48 hours. There was a non-significant rise in the percentage of lymphocytes over the same time period. No significant changes were seen in the percentage of columnar cells or total number of macrophages.
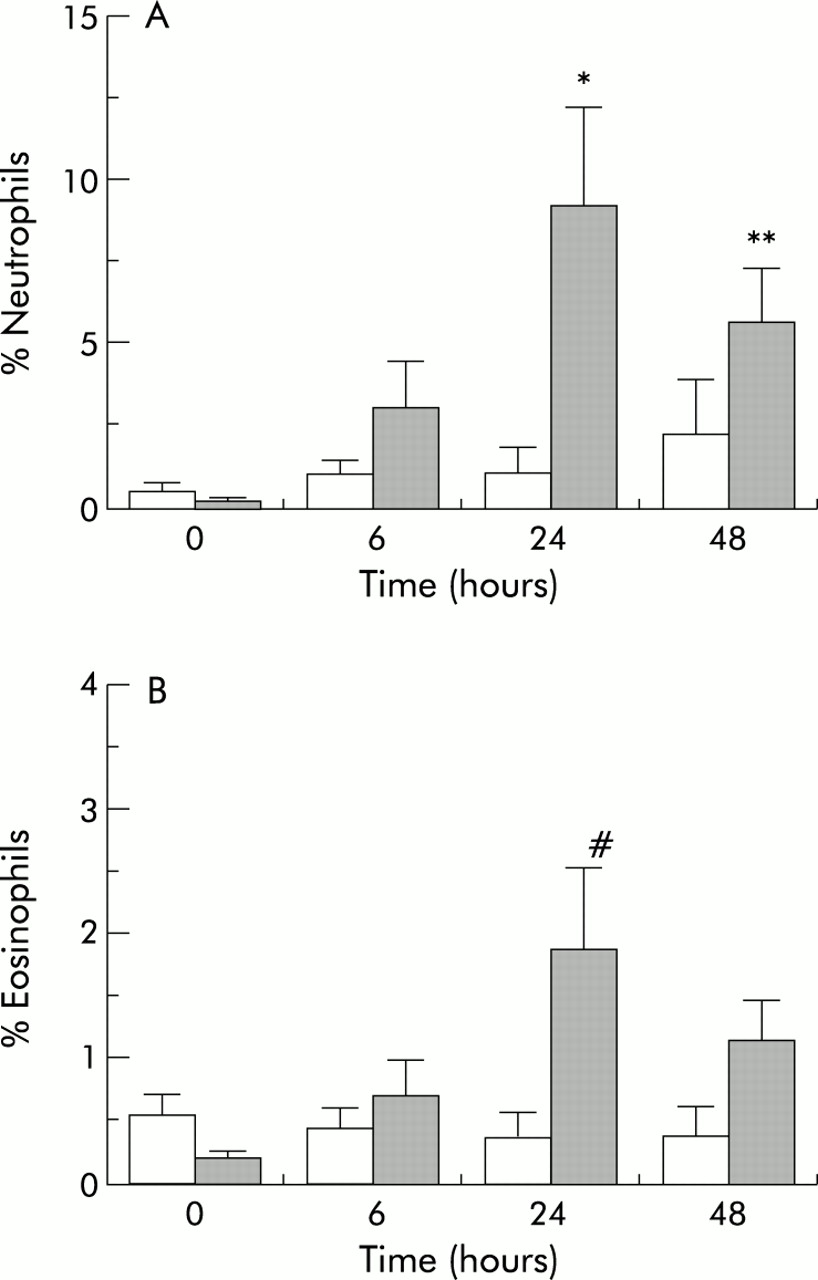
{kind=link}
{kind=link}
(A) Effect of control (open bars) or tumour necrosis factor alpha (TNFα, shaded bars) inhalation on change in percentage neutrophils from baseline for the 3 days of the study (ANOVA, p<0.01; post hoc Bonferroni corrected t test, *p<0.05,**p<0.02). (B) Effect of control or tumour necrosis factor alpha (TNFα) inhalation on change in percentage eosinophils from baseline for the 3 days of the study (ANOVA, p<0.05; post hoc Bonferroni corrected t test, #p=0.05).
Cytokine analysis
Messenger RNA from the sputum at 0, 6, 24 and 48 hours after rhTNFα inhalation showed an increase in IL-14, IL-15, and TNFα message compared with the control arm in the majority of subjects at 6 hours only and, although this did not achieve statistical significance (Wilcoxon sign rank test, data not shown), it would be the expected time at which other mediators would be induced. The remaining cytokines showed no increase in message above baseline.
Exhaled NO and CO
There was a non-significant rise in both eNO and eCO after both control and rhTNFα inhalation. Exhaled NO 24 hours after TNF inhalation, expressed as percentage of baseline, was 110.0% (95% CI 80.0 to 139.9) compared with 109.8% (95% CI 97.8 to 121.8) in controls; after 48 hours the corresponding values were 99.7% (95% CI 52.6 to 146.7) and 101.3% (95% CI 54.8 to 147.8), respectively.
Exhaled CO 24 hours after TNFα inhalation, expressed as percentage baseline, was 108.3% (95% CI 89.9–126.7) compared with 122.6% (95% CI 96.3 to 148.9) in controls; after 48 hours the corresponding values were 82.1% (95% CI 54.5 to 109.7) and 109.7% (95% CI 94.2 to 125.2), respectively.
DISCUSSION
TNFα is a multifunctional cytokine with a wide range of activities. It has been implicated in a number of diseases including asthma,5 rheumatoid arthritis, multiple sclerosis, and other inflammatory disorders. It is implicated in cell death and apoptosis but it is able also to generate a non-cytotoxic inflammatory response in certain situations. It has been shown conclusively to be released immediately from mast cells after encounter with specific allergens,2,19,20 and is therefore implicated in allergic asthma.
TNFα is associated with the local upregulation of the adhesion molecules E-selectin, VCAM-1, and ICAM-1 which are involved in leucocyte migration. Both VCAM-1 and ICAM-1 have been shown to be involved in the transmigration of granulocytes across endothelial cultures in vitro21–23 and increased VCAM-1 expression is associated with a preponderance of TNFα in eosinophilic nasal polyposis.24 TNFα appears to exert its effect on the increased migration of eosinophils and neutrophils through these adhesion factors, with increased eosinophil attachment reliant upon α4 integrin binding to the upregulated VCAM-1 expression23 and migration associated with upregulated ICAM-1 expression,22 although neutralising experiments have implicated the β2 integrins.25 TNFα supports GM-CSF mediated eosinophil migration, which is dose dependent for both cytokines, and this migration is inhibited by actinomycin D. Both TNFα and IL-1β increase ICAM-1 expression and antibodies to ICAM-1 inhibit this migration through human umbilical vein endothelial cells (HUVEC), and may therefore act in concert with IL-5 in this regard.22 In asthma, TNFα is more frequently expressed in the airways than in normal subjects, and in allergic polyposis this increase in TNFα is associated with an increase in VCAM-1 and eosinophilic infiltration.24,26 It has become apparent that, although eosinophil migration is classically thought to depend upon eotaxin, IL-4, IL-5, and GM-CSF, there is evidence now to suggest that TNFα can upregulate cells to express IL-527 and eotaxin.28
The migration of neutrophils into the airway as demonstrated by these experiments in asthmatic subjects confirms our previous findings in normal non-asthmatic subjects.9 This sputum neutrophilia is associated with an increase in airway reactivity and these findings would fit the hypothesis that airway inflammation induces increased airway reactivity. Sputum neutrophilia is seen in asthma, particularly fatal asthma,29–31 although it is recognised less often than the association of asthma with sputum eosinophilia. Sputum neutrophilia could be the result of TNFα release from allergen activated mast cells and macrophages, or via bacterial activation. It is now apparent that both neutrophils and eosinophils are associated with the late phase of the allergic response.32–34 Neutrophils are associated in asthma with increased migratory responses, superoxide generation, and increased bronchial ring contractility.35–37
Induced sputum mRNA suggested an increase in message for TNFα, IL-14, and IL-15. The roles of IL-14 and IL-15 have not yet been clarified in asthma, but it is becoming apparent that IL-15 has potent growth factor activity for mast cells in vitro38 and thus may be a mechanism for perpetuating the allergic inflammatory response. The finding that sputum TNFα mRNA was increased after TNFα exposure was unexpected as a negative feedback mechanism might have been anticipated. It is possible that, on exposure to a low dose of rhTNFα, there is an initial amplification of activation before inhibitory mechanisms come into play. It was likewise anticipated that IL-5 would be upregulated by the nebulised TNFα as it has been shown to upregulate IL-5 and downregulate IL-4.39 Neither change was seen but, again, it is difficult to know whether the local dose delivered in this study would have been comparable to other reports. The dose of rhTNFα used was, however, comparable to previous reports of total TNF isolated from BAL fluid after allergen challenge.7
Exhaled gas analysis failed to show any significant changes. This may have been because of the small number of study subjects, and there are no data in the literature to suggest an appropriate sample size but, importantly, it also could be masked by saline challenge for sputum induction. Sputum induction by itself will generate a neutrophil sputum leucocytosis at about 24 hours9 and this may be the mechanism whereby a slight increase in eNO and eCO is seen in this arm of the study. To show a separate effect of inhaled rhTNFα alone would require a different study design without the induction of sputum. TNFα has been described as upregulating inducible NO synthase40,41 and it would be expected that this might be reflected in an increase in eNO, but possibly a larger dose of nebulised TNFα would be required to demonstrate this effect.
In conclusion, it appears that small quantities of cytokines such as TNFα can have proinflammatory effects that are measurable in the airways of subjects with mild asthma. While glucocorticosteroids are the most effective means of inhibiting this inflammation, it is possible that, by inhibiting these cytokines directly, a reduction in airway inflammation and reactivity may be achieved and could be a useful method of reducing exposure to glucocorticosteroids.
Acknowledgments
This study was funded by the NH&MRC, Australia. The authors thank the subjects for donating their time.