Article Text

Abstract
Background: The use of reverse transcription-polymerase chain reaction (RT-PCR) to measure mRNA levels has led to the common use of β-actin and GAPDH housekeeping genes as denominators for comparison of samples. Expression of these genes is assumed to remain constant, so normalising for variations in processing and signal quantitation. However, it is well documented that β-actin and GAPDH expression is upregulated with proliferation, activation, and differentiation. We hypothesised that airway samples which differ in their cellular profiles and activation status have different levels of expression of GAPDH and β-actin.
Methods: The mRNA for β-actin, GAPDH, and interleukin (IL)-2 was measured in bronchoalveolar lavage (BAL) fluid cells and endobronchial biopsy tissue by competitive RT-PCR in a cross sectional study of 26 normal controls and 92 asthmatic subjects.
Results: For both BAL fluid cells and biopsy tissue, asthmatics overall had reduced expression of GAPDH and β-actin mRNA. In asthmatic subjects not using inhaled corticosteroids (ICS), GAPDH mRNA levels in both BAL fluid and biopsy tissue, and β-actin mRNA in BAL fluid cells were 10 times lower than samples from both normal controls and from asthmatic subjects using ICS. β-Actin mRNA in biopsy specimens showed the same pattern of expression, but asthmatic subjects not using ICS were not significantly different from those receiving ICS treatment. IL-2 mRNA levels did not differ between the subject or treatment groups but, when expressed as a ratio with β-actin, significant differences were seen.
Conclusions: β-Actin and GAPDH used as denominators of gene expression quantitation in asthma research can cause confounding. Housekeeping genes need careful validation before their use in such quantitative mRNA assays.
- housekeeping gene expression
- asthma
- inhaled corticosteroids
Statistics from Altmetric.com
Attempts to develop quantitative assays of mRNA in samples have led to the common use of housekeeping gene expression to normalise the data. Assays based on Northern blots and semiquantitative reverse transcription-polymerase chain reaction (RT-PCR) techniques are characterised by multiple steps, each introducing variation from sample to sample. In order to relate the results to each other and to the samples from which they originated, molecular biology assays need endogenous control(s), which are similarly influenced by the efficiency of each step.1
Housekeeping genes are defined by specific gene promoter elements which determine that they are expressed constitutively in every cell, but that does not necessarily mean that their expression is not regulated.2 However, the use of housekeeping genes in molecular biology assays relies on the assumption that their levels of expression remain the same from cell to cell, sample to sample, treatment to treatment, and/or patient to patient. While all methodologies need validation, it is difficult to determine whether housekeeping gene expression has biological variation in the absence of a validated alternative means of normalising the data. This is particularly so if housekeeping gene expression is the sole arbiter of the assay.
β-Actin, a cytoskeletal protein, and GAPDH, an enzyme of glycolysis, are the two most widely used housekeeping genes,1 on the basis of rather anecdotal evidence of their consistency of expression. Quantitative RT-PCR assays based on competition with a DNA template sharing primer sequences with the mRNA of interest have recently been applied to lung samples.3,4 We have used such a reverse transcription-competitive PCR method, not itself dependent on housekeeping gene expression, to measure β-actin and GAPDH mRNA levels directly in a cross sectional series of biopsy tissue and BAL fluid cells from normal controls and subjects with asthma.
METHODS
Subjects
Ninety two asthmatic volunteers took part in the study. Airway endobronchial biopsy samples were obtained from 70 patients and BAL fluid cells from 75 with matching samples being obtained from 53 patients (table 1). The asthmatic subjects fulfilled the American Thoracic Society criteria5 for asthma with documented airway reversibility to inhaled bronchodilator and increased responsiveness to methacholine. They all used albuterol for relief of symptoms as needed, with 79 patients using regular inhaled corticosteroids (ICS). Salmeterol (50 μg twice daily) was used by 12 patients from whom BAL fluid cells were obtained and in five from whom biopsy specimens were taken. No patient was taking any other anti-asthma medication. Volunteers who had suffered from acute respiratory tract infection during the previous 4 weeks, had any change in asthma medication, or been admitted to hospital with airway disease in the 4 weeks preceding the study were excluded.
Bronchoscopy subject profiles
Twenty six normal volunteer control subjects were also recruited. They had no history of asthma or other respiratory diseases, were using no medication, had no bronchodilator response to albuterol, and a negative challenge test to 3.2 mg inhaled methacholine. All asthmatic and control subjects were non-smokers and gave informed written consent before commencing the study, which was approved by the Alfred Hospital ethics committee.
Atopic status, spirometric measurements, and bronchial responsiveness to methacholine were determined as previously described.6 Bronchoscopy was performed within 2 days of methacholine challenge. Samples were transported on ice to the laboratory and two biopsy specimens were snap frozen in liquid nitrogen within 15 minutes and stored at –80°C. BAL fluid cells in aliquots of 1–4 × 106 cells were pelleted by centrifugation at 1200 rpm for 15 minutes, snap frozen, and stored at –80°C.
RNA extraction
RNA from BAL fluid cells and endobronchial biopsy tissue was extracted with RNeasy Total RNA Kit (Qiagen, Hilden, Germany). Tissue was homogenised in 700 μl lysis buffer RLT using a Polytron rotor-stator (Kinematica, Luzern, Switzerland) followed by aspiration 10 times through a 21 gauge syringe needle to shear chromosomal DNA. Cell pellets were resuspended in 700 μl lysis buffer RLT by aspiration 10 times through a 21 gauge syringe needle. RNA was eluted from spin columns with 40 μl diethylpyrocarbonate treated water. Aliquots of 4 μl RNA were quantitated by capillary spectrophotometry using an absorbance capillary adaptor cell (Helix, San Diego, CA, USA) and a Cary 1 spectrophotometer (Varian, Melbourne, VIC, Australia) with background correction at 320 nm. Samples with (A260–A320)/(A280–A320) ratios less than 1.7 and/or yields less than 0.5 μg total RNA were excluded from subsequent analysis.
Competitor templates and primers
DNA competitor templates for β-actin and GAPDH PCR were obtained from Clontech (Palo Alto, CA, USA). IL-2 PCR used a synthetic mRNA competitor, pAW109 cRNA (Perkin-Elmer, Foster City, CA, USA). GAPDH PCR used forward primer TGA AGG TCG GAG TCA ACG GAT TTG GT and reverse primer CAT GTG GGC CAT GAG GTC CAC CAC (synthesised by GeneWorks, Adelaide, SA, Australia), producing competitor products of 630 bp and native products of 983 bp. The β-actin primers were forward primer GCA CCA CAC CTT CTA CAA TG and reverse primer TGC TTG CTG ATC CAC ATC TG. β-Actin PCR products were 619 bp from competitor and 838 bp from cDNA. IL-2 forward primer, GAA TGG AAT TAA TAA TTA CAA GAA TCC, and reverse primer, TGT TTC AGA TCC CTT TAG TTC CAG, produce products of 229 bp from mRNA and 305 bp from cRNA. All competitors were diluted to working concentrations with 50 μg/ml nuclease-free glycogen (Roche Diagnostics Australia, Castle Hill, NSW, Australia) in 10 mM Tris HCl, pH 7.5.
Validation of competitive PCR
To establish that mRNA and competitor templates amplify equally, a “changing input-output ratio” series of reactions was set up for each gene. For GAPDH and β-actin, aliquots of BAL cell cDNA were combined in 100 μl PCR with a serial titration of competitor DNA from 8 × 105 to 4 × 102 copies. Reactions were split into duplicate 50 μl aliquots and amplified for 40 cycles. To test the validity of IL-2 RT-PCR, a serial titration of pAW109 cRNA from 2 × 103 to 5.12 × 105 copies per reaction was established in 15 μl cDNA synthesis reactions containing 180 ng PHA stimulated PBMC (prepared as previously described7) RNA. Aliquots of 2 μl cDNA were added to 100 μl PCR which were then split into duplicates and amplified for 35 and 40 cycles.
Reverse transcription and competitive PCR
A pilot study of a small number of BAL cell samples and biopsy samples was conducted to optimise the amount of competitor to use in subsequent reactions. For β-actin and GAPDH the amount of competitor was adjusted so that the 100% and 98%, respectively, of reactions gave ratios greater than 0.1.
cDNA was synthesised from up to 2.85 μg RNA in a volume of 57 μl consisting of 1× GeneAmp PCR buffer, 5 mM MgCl2, 1 mM deoxyribonucleotide triphosphates, 0.8 U/μl RNase inhibitor, 2 μM Oligo d(T)16 and 2 U/μl MuLV Reverse Transcriptase (all from Perkin-Elmer). For cDNA synthesis from BAL cell RNA, 3 × 105 copies of pAW109 cRNA were added to each reaction, while for biopsy RNA, 4 × 103 copies of pAW109 RNA were used. Reactions were incubated for 1 minute at 15°C and the temperature was then increased to 42°C over 9 minutes, followed by 1 hour at 42°C, 5 minutes at 85°C, and 1 minute at 4°C.
For GAPDH and β-actin competitive PCR, 2 μl of 1 in 80 diluted cDNA was added to a 50 μl reaction consisting of 1 U AmpliTaq Gold, 1 × GeneAmp PCR buffer, 1.5 mM MgCl2, 200 μM deoxyribonucleotide triphosphates (all from Perkin-Elmer), 0.2 μM forward and reverse primers. GAPDH competitor was added at 5 × 103 and 2.5 × 104 copies to PCR on biopsy tissue and BAL cells, respectively. β-Actin PCR was conducted with 4 × 103 and 1 × 105 copies of competitor for biopsy tissue and BAL fluid cells, respectively. Thermal cycling was conducted in an MJ Research PTC-200 (Watertown, MA, USA), with the GAPDH temperature profile commencing with an initial denaturation of 8 minutes at 94°C followed by 40 cycles of 94°C for 1 minute, 64°C for 1 minute, and 72°C for 1 minute, concluding with 7 minutes at 72°C and 1 minute at 4°C. β-Actin PCR was conducted similarly but the annealing temperature was 60°C. IL-2 PCR was set up as above except that 5 μl of cDNA was used and amplification was carried out with an annealing temperature of 56°C for 45 cycles.
The PCR products were resolved by electrophoresis through 1.5 mm, 4% 19:1 PAGE gels (acrylamide:bisacrylamide; Bio-Rad Laboratories, Hercules, CA, USA) in 2× TAE buffer7 using active cooling to maintain a temperature of 16–18°C. PAGE gels were stained by overlay with 1:10 000 SYBR Green I (Molecular Probes, Eugene, OR, USA) in 2× TAE for 45 minutes and then scanned directly in a laser based fluorescence scanner (FluorImager 575, Molecular Dynamics, Sunnyvale, CA, USA). PCR product band volumes were quantitated with ImageQuaNT software (Molecular Dynamics). Native/competitor product ratios were calculated and the levels of gene expression reported as copies of mRNA per μg total RNA.
Data analysis
The Pearson correlation was used to test associations between GAPDH and β-actin and between sample types. The differences between controls and asthmatic subjects using and not using ICS were analysed using a generalised linear model, with an interaction between asthma and ICS use being fitted to the model. The confounding variables used in the modelling as appropriate included age, sex, atopic status, ICS usage, use of long acting β2 agonists, and RT-PCR batch factors. The results are presented as least squares mean (SE) values. The Mann-Whitney U test was used for intergroup comparisons for IL-2/β-actin ratios. Statistical analyses were performed using SAS version 6.12 software (SAS Institute, Cary, NC, USA).8
RESULTS
Validation of competitive PCR
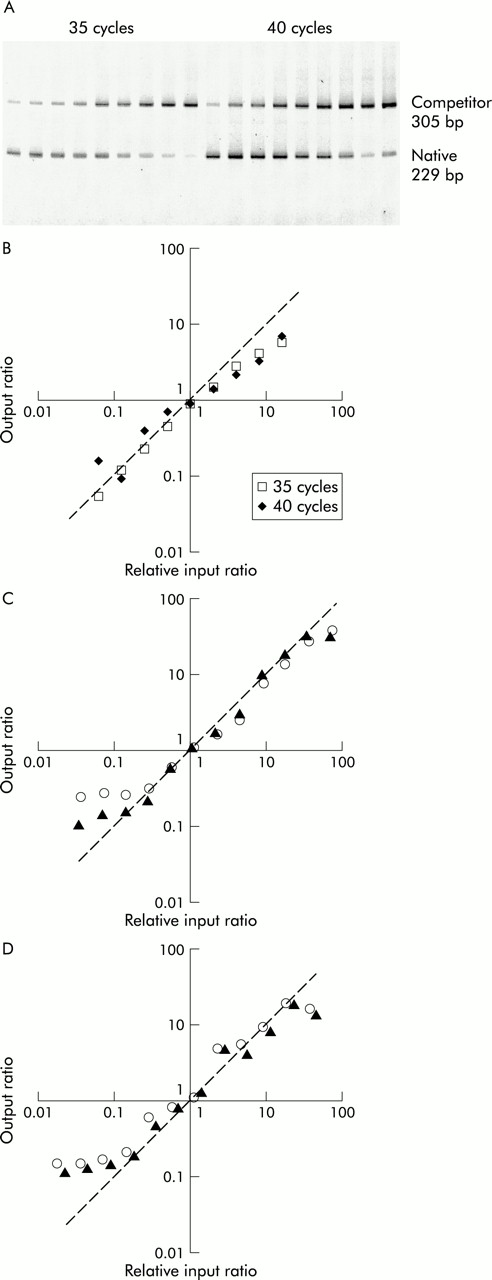
Titration of each housekeeping gene competitor against constant aliquots of BAL fluid cell cDNA in changing ratio experiments showed that, in each case, the ratios followed the predicted line for output ratios greater than about 0.1 (fig 1). Similarly, serial titration of the IL-2 competitor pAW109 cRNA against PHA stimulated PBMC RNA followed by subsequent RT-PCR produced output ratios that changed in proportion to the input ratios. Thus, GAPDH, β-actin, and IL-2 PCR products competed equally with their respective competitor templates, allowing the input copies of mRNA to be calculated.

Test of equal amplification efficiency of competitive PCR by titration of competitor. (A) PAGE of IL-2 competitive RT-PCR of changing input ratio series with increasing amount of competitor left to right. (B)–(D) The output native/competitor ratios of each series is plotted against input ratios relative to the one or two output ratios closest to 1. The actual data lie close to the line of prediction where output ratios equal input ratios. The line of prediction is indicated by a dashed line with a slope of 1. (B) Duplicate IL-2 RT-PCR were conducted for 35 and 40 cycles. β-actin (C) and GAPDH (D) titrations were replicated (opened and closed objects) at 40 cycles.
GAPDH and β-actin gene expression
GAPDH and β-actin mRNA levels were significantly correlated in both biopsy tissue (r2=0.48, n=85, p=0.0001) and BAL fluid cells (r2=0.51, n=97, p=0.0001; fig 2A and B). The correlation coefficients indicate that only half of the variability of one is explained by the other, suggesting that at the resolution of this methodology GAPDH and β-actin mRNA levels are not equivalent. Furthermore, GAPDH gene expression was more variable in biopsy samples and was approximately five times lower in BAL fluid cells than β-actin expression.

Pearson correlations between GAPDH and β-actin gene expression in RNA from (A) BAL fluid cells and (B) biopsy tissue.
Cross-sectional comparisons of gene expression
The gene expression of β-actin and GAPDH were compared with respect to atopic status, asthmatic status, and ICS use. GAPDH gene expression in biopsy specimens from asthmatic subjects not using ICS (least squares mean (SE) 8.1 (1.3) × 105 mRNA copies/μg RNA) was significantly lower than in biopsy specimens from normal controls (1.1 (0.1) × 107 mRNA copies/μg RNA, median difference 1.0 × 107, 95% confidence interval (CI) 0.68 to 1.4 × 107, p=0.0001) and from asthmatic subjects using ICS (1.2 (0.2) × 107 mRNA copies/μg RNA, median difference 1.1 × 107, 95% CI 0.71 to 1.5 × 107, p= 0.0001, fig 3A). Asthmatic subjects as a group, whether taking ICS or not, were significantly different from controls (7.1 (1.9) × 106 mRNA copies/μg RNA, median difference 3.9 × 106, 95% CI 1.9 to 5.9 × 106, p=0.0003). The BAL cell levels of β-actin and GAPDH mRNA showed similar differences between groups as the biopsy GAPDH mRNA levels, with corresponding p values (fig 3B and D). These differences in gene expression are in contrast to the profile of cell types in BAL fluid which did not differ significantly between the patient groups (table 2).
Profiles of major cell types in BAL fluid cell samples*
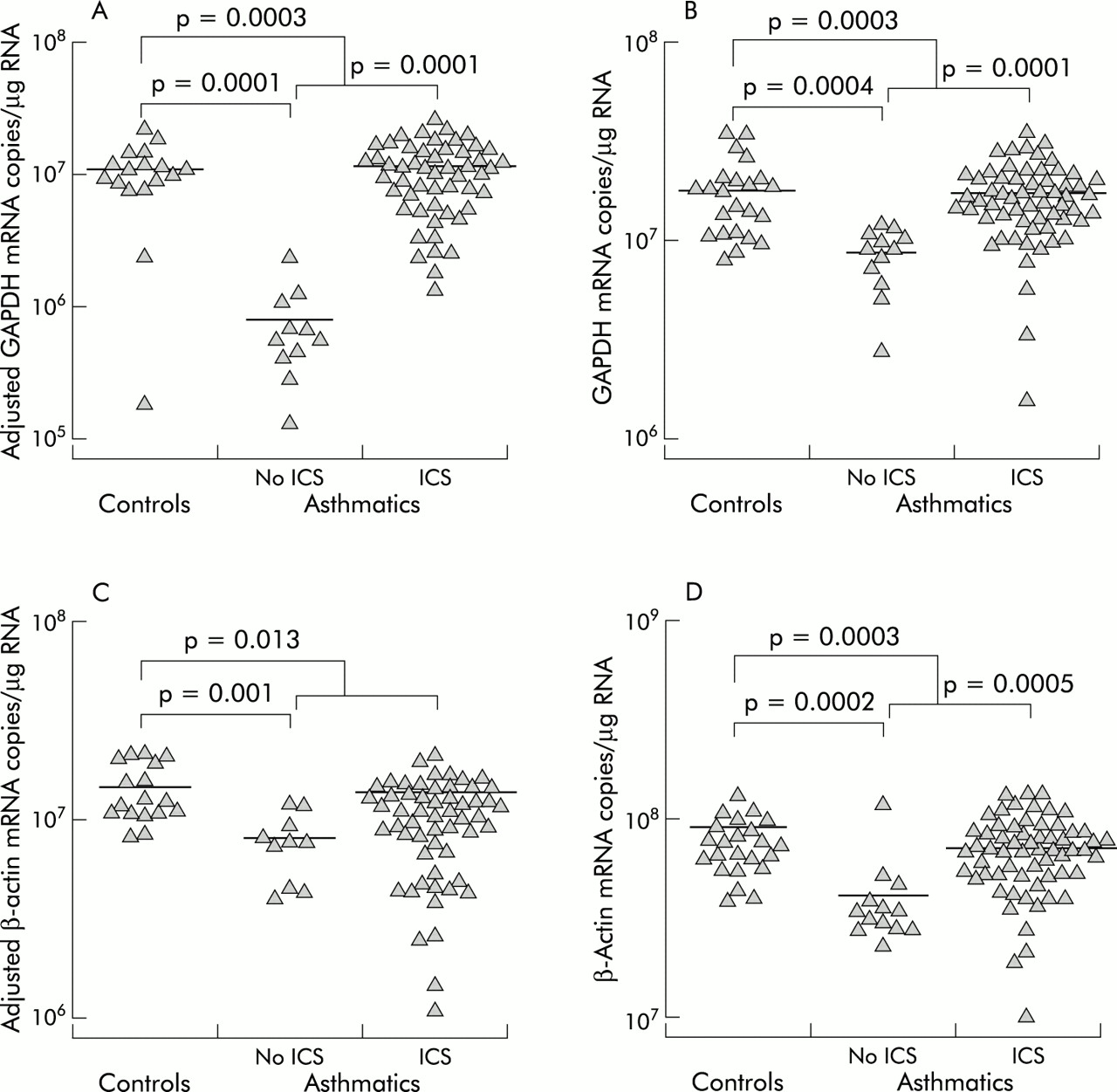
Cross sectional comparisons of GAPDH and β-actin gene expression. GAPDH gene expression in (A) biopsy tissue and (B) BAL fluid cells. β-actin mRNA levels in (C) biopsy tissue and (D) BAL fluid cells. The mRNA levels in biopsy tissue have been corrected for contamination with pseudogene sequences. The small differences between the number of data points in the figures and the number of samples in table 1 is due to the small percentage of expected failed reactions. Failed reactions were not repeated in order to reduce the potential for systematic bias.
β-Actin mRNA expression in biopsy tissue differed in that asthmatics taking ICS were similar to those not on ICS (p=0.26, fig 3C). However, in common with the other comparisons, normal controls (1.5 (0.1) × 107 mRNA copies/μg RNA) had higher β-actin mRNA levels than asthmatic subjects not taking ICS (8.1 (1.4) × 106 mRNA copies/μg RNA, median difference 6.9 × 106, 95% CI 3.4 to 10 × 106, p=0.001) and higher levels than all asthmatics (9.3 (1.5) × 106 mRNA copies/μg RNA, median difference 5.7 × 106, 95% CI 1.4 to 10 × 106, p=0.013).
Effect of using housekeeping genes as denominators
To demonstrate the effect of using β-actin and GAPDH as denominators of mRNA expression for genes of interest, IL-2 competitive PCR was conducted on BAL cell RNA using the same cDNA reaction as for β-actin and GAPDH. IL-2 mRNA levels in BAL cells did not differ between normal controls (4.6 (0.9) × 104 copies/μg RNA), asthmatics not using ICS (4.8 (1.2) × 104 copies/μg RNA), and asthmatics taking ICS (3.0 (0.7) × 104 copies/μg RNA; fig 4). However, when IL-2 mRNA levels were expressed as a ratio with β-actin there were significant differences, with asthmatics not using ICS having a higher ratio (1.2 (0.4) × 10–3) than normal controls (6.2 (1.8) × 10–4, median difference 9.24 × 10–4, 95% CI 0.95 to 21.0 × 10–4, p=0.03) and asthmatics using ICS (4.5 (0.9) × 10–4, median difference 1.1 × 10–3, 95% CI 0.3 to 2.1 × 10–3, p=0.003). Thus, apparent differences in the IL-2/β-actin ratios are confounded by the differences in β-actin mRNA levels between the groups (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of using β-actin as a denominator of IL-2 BAL fluid cell mRNA levels. To compare IL-2 and β-actin mRNA levels and their ratios, the subject groups least squares means are presented as percentages of the respective normal control least squares mean. Error bars are given as percentage SE. *Significant differences were observed for β-actin (as in fig 3D), and IL-2/β-actin ratios in asthmatic subjects not using inhaled corticosteroids (ICS) are significantly different from normal controls (p=0.03) and asthmatic subjects using ICS (p=0.003).
DISCUSSION
This study is the first systematic analysis of the validity of using housekeeping gene expression to normalise mRNA quantitation in lung samples. We have used reverse transcription followed by competitive PCR to quantitate β-actin and GAPDH gene expression in airway biopsy tissue and BAL fluid cells from normal controls and asthmatic patients. Asthmatics as a group and, in particular, asthmatics not using ICS, were found to have decreased expression of both β-actin and GAPDH. Quantitation of mRNA in asthma will be confounded by these differences if GAPDH and β-actin are used to normalise data, and this was shown for IL-2 mRNA levels expressed as ratios with β-actin.
To develop a technique of gene expression measurement that would be independent of housekeeping gene expression, we sought to maximise the quantitative nature of each step. Capillary spectrophotometry with background correction was used to analyse the sample RNA accurately so that a consistent quantity and quality was added to the assay. The competitive nature of the PCR and the subsequent direct quantitation by scanning fluorescence controls for the latter part of the assay. The only step without endogenous control is the cDNA synthesis.
Nevertheless, the assay is still highly controlled and we have assumed that there is similar reverse transcription efficiency between subject groups. To prevent systematic bias in efficiency affecting comparisons, the RNA samples were stratified according to RNA concentration before cDNA synthesis. We therefore believe that it is very unlikely that the differences in GAPDH and β-actin gene expression with asthma were due to the RNA from ICS untreated asthmatic samples having been transcribed with 10 times less efficiency.
Analyses of correlations between GAPDH and β-actin and between biopsies and BAL cells suggest that the two housekeeping genes vary independently of each other. A single cDNA synthesis reaction was conducted for each sample so that β-actin and GAPDH could be compared without confounding by differences in reverse transcription efficiency. In addition, a limited number of samples taken 3 months apart from clinically stable asthmatics showed a complete lack of concordance in β-actin and GAPDH mRNA levels (data not shown). Normalisation of a gene of interest by housekeeping gene expression will incorporate the biological variability of the housekeeping gene and so add to assay variability and decrease the resolution of the method.
In a rat model of particulate induced lung injury, RNA degradation has been observed concomitant with a time dependent influx of eosinophils in inflamed pulmonary tissue.9 The degraded RNA samples produced significantly reduced β-actin levels in an RT-PCR assay. Asthmatics not using ICS will not have eosinophilic inflammation as severe as that generated by such an animal model, but increased eosinophil numbers and/or activation correlating with disease severity is a well documented feature of asthma.10 The asthmatic subjects in this study were clinically stable and the percentage of eosinophils in the BAL fluid samples were not significantly different between the subject groups (table 2), making this potential source of confounding unlikely.
Our results could be interpreted as showing, not that the mRNA levels of β-actin and GAPDH per cell differed between normal controls and asthmatics, but that the total RNA pool was increased with asthma. Ribosomal RNA comprises more than 90% of the total cellular RNA measured by spectrophotometry and varies with cell type, functional state of the cell, and stage of the cell cycle.11 With normalisation to total RNA, differences in the proportion of mRNA in total RNA between patient groups could confound the quantitation of gene expression. However, in the BAL cell samples there were no significant differences between the groups in cell types (table 2), nor were there any significant correlations between housekeeping gene mRNA levels and the percentage of the major cell populations of macrophages, lymphocytes, or eosinophils. Conversely, there was a substantial difference in cellular composition between biopsy and BAL cell samples, but the same pattern of housekeeping gene expression with asthma and ICS use was found. There seems little chance that the tenfold difference in housekeeping gene mRNA levels between asthmatics with and without ICS treatment could be due to effects on total RNA composition.
While molecular biologists have tended to regard GAPDH and β-actin as simple housekeeping genes, evidence has been growing over the past decade which indicates that, while constitutively expressed, their rate of transcription is influenced by a number of factors. Both β-actin and GAPDH mRNA levels vary with cellular proliferation12–16 and their transcription is upregulated rapidly in response to mitogenic stimuli including epidermal growth factor, transforming growth factor-β (TGF-β), and platelet derived growth factor.17–19 In view of this, it is ironic that both β-actin and GAPDH have been used to normalise the quantitation of the mRNA of growth factors such as TGF-β, including in some studies in asthma.20,21
The observed variability of expression of β-actin and GAPDH genes emphasises their important physiological roles. The β-actin cytoskeleton functions in cellular shape and anchorage where transmembrane glycoproteins link fibronectin in the extracellular matrix with actin microfilaments on the cytoplasmic side of the membrane.22 It is not surprising therefore that cellular proliferation, activation, and/or differentiation induces upregulation of β-actin gene expression and subsequent cytoskeletal remodelling.22,23 Similarly, GAPDH expression would be expected to vary as it has a diverse range of functions unrelated to its glycolytic activity. GAPDH has been shown to be involved in membrane transport and membrane fusion, microtubule assembly, nuclear RNA export, protein phosphotransferase/kinase reactions, the translational control of gene expression, DNA replication, and DNA repair.24 Indeed, these functions of β-actin and GAPDH determine the need, not only for their expression in every cell, but also confer the requirement for tightly regulated expression.
In spite of the growing body of evidence of differential regulation of GAPDH and β-actin, these housekeeping genes have been widely used in mRNA quantitation for three main reasons. Firstly, there has been a substantial lack of validation studies to discredit the methodological concept. Secondly, control of RNA quality and cDNA synthesis efficiency without housekeeping gene expression is otherwise difficult. Alternatives include incorporation of radioactive labels during cDNA synthesis25 and co-RT-PCR of synthetic mRNA exogenous standards.26 Lastly, the use of housekeeping gene expression has allowed relatively inexpensive and simple technology to be used. Often the justification for using semiquantitative methods over quantitative assays has been the large differences expected to be found, therefore only requiring a low resolution technique. However, the differences detected here in housekeeping gene expression question this justification.
The advent of real time quantitation of PCR has circumvented the problems associated with controlling PCR amplification and detection.27,28 However, normalising samples for RNA quality and reverse transcription efficiency remain as potential sources of confounding of data. Clearly, mRNA assays need to be designed to allow validation of the internal standards being used and each new application of a given method should include an analysis of consistency of expression.
Acknowledgments
The authors thank Ros Bish, Sally Gollant, Dr Frank Thien, Dr Xun Li, and Dr David Reid for their clinical assistance and Bernadette Orsida, Tiffany Bamford, Michael Pais and Bryce Feltis for sample reception. This work was supported by GlaxoWellcome Australia and the NHMRC, Australia.