Article Text

Abstract
Genetic factors including α1-antitrypsin deficiency are important in COPD. Candidate gene association studies in COPD are reviewed. Efforts to identify interactions between genetic factors and environmental determinants such as smoking may lead to improved understanding of the pathogenesis of the disease.
- chronic obstructive pulmonary disease
- genotype-environment interaction
- genetics
- smoking
Statistics from Altmetric.com
Chronic obstructive pulmonary disease (COPD) is a complex disease which is influenced by genetic factors, environmental influences, and genotype–environment interactions. Genotype–environment interactions have been of interest to geneticists for decades. In 1946 Haldane noted that “the interaction of nature and nurture is one of the central problems in genetics”, and it remains so today.1 A genotype–environment interaction can be defined as a non-additive contribution of genetic and environmental factors to the expression of a phenotype.2 Thus, the presence of both genetic and environmental influences on a phenotype does not necessarily imply a genotype–environment interaction; non-additivity of these influences must be present, and non-additivity is observed on a certain scale of measurement. The scale of measurement of clinical variables may not correspond to the scale on which genetic and environmental factors act. Transformations of scale (e.g. logarithmic) may remove genotype–environment interactions, which can be useful for analytical approaches that assume that no genotype–environment interaction is present.3
It is not necessary to know the specific genetic and environmental influences on a phenotype to determine if a genotype–environment interaction is present. Many agricultural studies have compared crop strains in different geographical sites, providing evidence for a genotype–environment interaction without identifying the specific genetic or environmental differences involved.4 Of course, identification of the specific genes and the specific environmental factors that interact is of great interest in human genetics, and progress in the human genome project is making the identification of such interactions possible.
Cigarette smoking is clearly the major environmental determinant of COPD; the ability to quantify exposure to cigarette smoke with reasonable accuracy using a questionnaire provides unique opportunities for the study of genotype–environment interactions. For many other complex human disorders, environmental factors are probably important but are difficult to measure. In addition to cigarette smoking, a number of other environmental factors may also be important in the development of COPD. Recent work has focused on latent adenoviral infections as a potential environmental trigger for COPD5; in addition, childhood respiratory infections, air pollution, and occupational exposures have been suggested as environmental determinants.6 In this review we will discuss the genetics of α1-antitrypsin (AAT) deficiency, familial aggregation of COPD unrelated to AAT deficiency, and association studies of candidate genes in COPD in the context of genotype–environment interactions.
SEVERE AAT DEFICIENCY
Severe AAT deficiency usually results from a PI ZZ or PI Znull genotype, which is typically diagnosed as a PI Z phenotype by isoelectric focusing of serum. PI Z is frequently associated with the development of early onset COPD. Several large series of AAT deficient individuals have clearly shown that PI Z subjects who smoke cigarettes tend to develop more severe pulmonary impairment at an earlier age than non-smoking PI Z individuals.7–9 In addition, a follow up study of participants in the Swedish AAT Deficiency Register has shown that the rate of decline in forced expiratory volume in 1 second (FEV1) is significantly higher in PI Z current smokers than in never smokers or ex-smokers.10
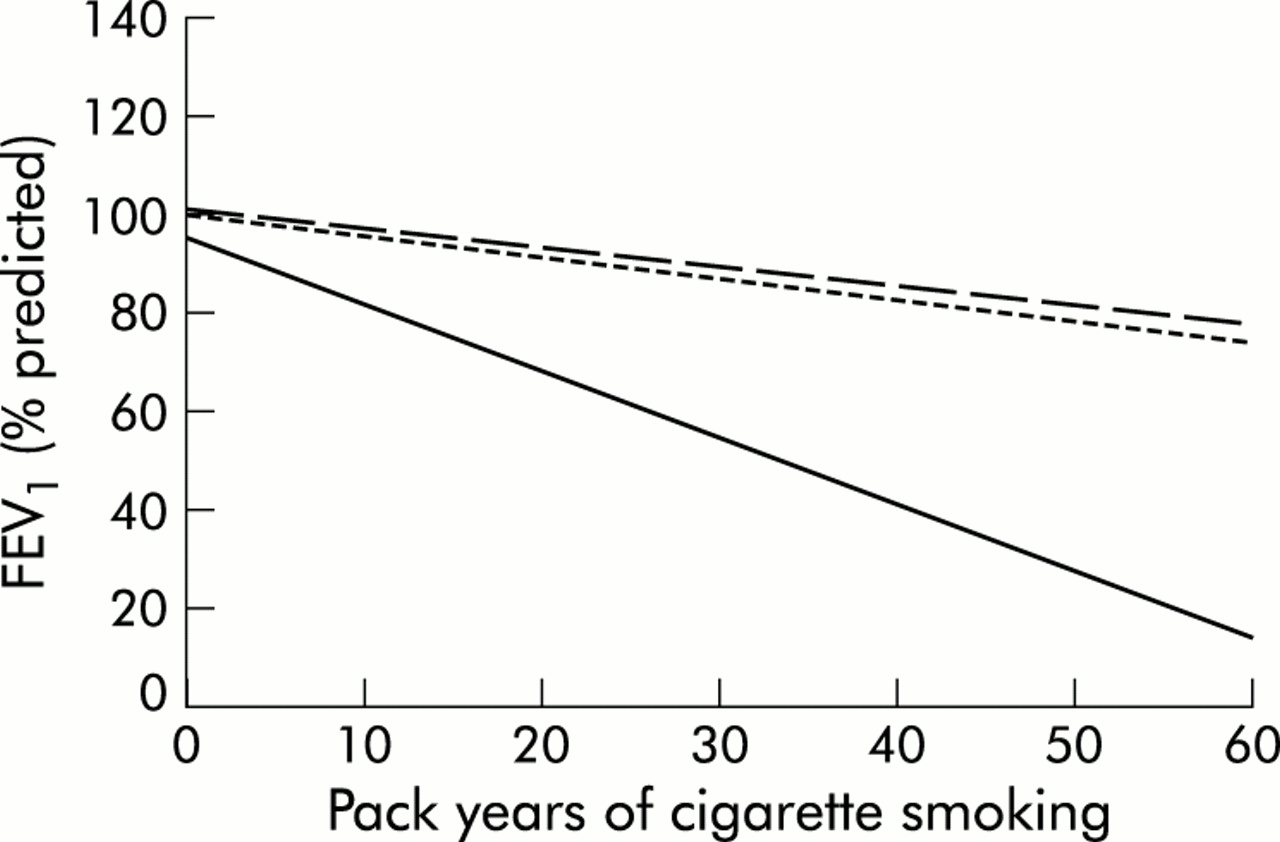
A significant fraction of the variability in pulmonary function among PI Z individuals is certainly explained by cigarette smoking; however, the development of COPD in PI Z subjects, even among current or ex-smokers, is not absolute. Silverman and colleagues11 studied 52 PI Z subjects and found that index PI Z subjects, who were tested for AAT deficiency because they had COPD and who were the first PI Z identified in their family, all had significantly reduced FEV1 values. Many non-index PI Z subjects, who were ascertained by various other means including family studies, population screening, and liver disease, had preserved pulmonary function. By comparing the slopes of the relationship between FEV1 and pack years of smoking in PI Z, PI MZ, and PI M subjects, Silverman and colleagues showed that there was a significant genotype–environment interaction between PI type and pack years of smoking (fig 1).12 However, the PI × smoking interaction was influenced by ascertainment; if only index PI Z subjects were included, all of whom had profoundly reduced pulmonary function, the greater sensitivity of PI Z subjects to increased smoking intensity could not be shown.

FEV1 as a function of pack years of smoking in non-index PI Z subjects (solid line), PI MZ subjects (dotted line), and PI M subjects (dashed line) in the Washington University AAT study. The steeper decline in FEV1 in response to pack years in PI Z subjects compared with PI M and PI MZ subjects indicates a significant genotype–environment interaction. Adapted from Silverman et al.12
With subjects identified from the Danish AAT Register, Seersholm and colleagues also found significantly higher FEV1 values in non-index PI Z subjects (not identified because they had COPD) than in index PI Z subjects (identified because they had COPD), despite similar ages and smoking histories.13 Moreover, Seersholm et al showed that index PI Z subjects had markedly reduced survival compared with non-index PI Z subjects.14
Few studies have considered whether factors other than smoking influence the development of lung disease. Black and Kueppers15 studied 54 PI Z individuals and found significant variability in pulmonary function and clinical symptoms, especially among non-smoking PI Z individuals. Piitulainen et al16 studied more than 200 non-smoking PI Z subjects from the Swedish AAT Deficiency Register, so they were able to examine the impact of risk factors for reduced pulmonary function without confounding by smoking. Not surprisingly, increasing age was associated with reduced pulmonary function in these non-smoking PI Z subjects. Male sex, wheezing symptoms, and occupational exposures to gas, fumes, or dust were also associated with reduced pulmonary function. Subsequent analyses in this population suggested that agricultural occupation and use of a kerosene heater were associated with reduced FEV1 levels; however, the generalisability of these findings may be limited since only a small number of PI Z subjects had these exposures.17
Mayer and colleagues recently assessed occupational exposures as a potential contributor to variable expression of lung disease in 128 PI Z subjects.18 They found that high mineral dust exposure was associated with reduced FEV1 and chronic cough, independent of smoking history. Although genotype–environment interactions have not been formally demonstrated, occupational and other environmental exposures may also be important determinants of the development of lung disease in PI Z subjects.
ROLE OF OTHER GENETIC FACTORS IN COPD
Familial aggregation of spirometric parameters and COPD
Studies of pulmonary function measurements performed in the general population and in twins have suggested that genetic factors influence variation in pulmonary function. Redline and colleagues assessed spirometric parameters in twins who were not selected for respiratory problems.19 For FEV1, a higher correlation in monozygotic twins (0.72) than in dizygotic twins (0.27) suggested that genetic factors influence variation in pulmonary function. Although such general population studies suggest that genetic factors influence pulmonary function, they do not address whether genetic factors influence the development of chronic airflow obstruction.
However, studies in relatives of patients with COPD have supported a role for genetic factors in the disease. Several studies in the 1970s and 1980s reported higher rates of airflow obstruction in first degree relatives of patients with COPD than in control subjects.20–22 For example, Kueppers et al found that the mean FEV1 in siblings of patients with COPD (90% of predicted) was significantly lower than in the siblings of matched control subjects (103% of predicted).23 Additional studies have shown familial aggregation for chronic bronchitis.24,25
More recently, Silverman and colleagues have focused on subjects with severe early onset COPD unrelated to severe AAT deficiency.26 Probands in this study had FEV1 <40% predicted at age <53 years. Among first degree relatives of early onset COPD probands, highly significant differences in FEV1 and FEV1/FVC were found when current or ex-smoking first degree relatives of early onset COPD probands were compared with control subjects. No significant differences in FEV1 or FEV1/FVC were found when lifelong non-smoking first degree relatives of early COPD probands were compared with lifelong non-smoking control subjects. This pattern of smoking related susceptibility, which was confirmed by multivariate analysis adjusting for age and pack years of smoking, would be consistent with genetic risk factors which interact with cigarette smoking to result in COPD. A similar pattern of smoking related susceptibility was also seen for chronic bronchitis and bronchodilator responsiveness.26,27
Segregation analysis and linkage analysis of pulmonary function and COPD
Segregation analysis, a statistical genetic method that determines whether the pattern of transmission of a phenotype within families is consistent with a major gene effect, has been applied to pulmonary function measurements in several studies. However, the results of these studies have been inconsistent. Using segregation analysis, Rybicki and colleagues28 found that a major gene influencing FEV1 was suggested in 85 COPD families but not in 56 non-COPD families. In segregation analysis of 455 randomly ascertained families from the NHLBI Family Heart Study, Wilk et al29 found evidence for a dominant major gene influencing FEV1. However, Chen et al30 performed segregation analysis of FEV1 in 214 randomly selected families and found no evidence for a major gene influence. Similarly, Givelber and colleagues found no evidence for a major gene influence on FEV1 in segregation analysis of 1408 families from the Framingham Study.31
Linkage studies have only recently been studied in COPD.31a,31b The absence of an accepted genetic model for the transmission of COPD from segregation analysis suggests that non-parametric linkage analysis methods will probably be required for COPD related phenotypes. Progress in linkage analysis may provide useful guidance for the selection of candidate genes for association studies.
ASSOCIATION STUDIES OF CANDIDATE GENES
Proteases
Heterozygotes for PI deficiency alleles
The issue of whether heterozygotes for PI deficiency alleles are at increased risk for the development of COPD is contentious. PI MS and PI MZ heterozygotes have reductions in serum AAT levels to ∼80% and 60% of normal, respectively. PI SZ compound heterozygotes are rare but have levels even lower at ∼40% of normal. PI SZ individuals (n=50) were shown to have “similar” levels of airflow obstruction to PI Z individuals, but only if they were smokers.32 In contrast, the frequency of the PI SZ genotype in 702 patients with COPD was not significantly different from that in the general population.33
Several case-control studies have shown an increased prevalence of PI MZ heterozygotes in patients with COPD compared with controls.34 In one of the largest studies, 526 German patients with COPD were compared with two control populations from the same country.35 There was a significant increase in the frequency of PI MZ heterozygotes in the patients (6%) compared with controls (1%), yielding an odds ratio (OR) of 5. In other studies the cases and controls were often not selected from the same population, raising concerns of spurious associations due to population stratification.
An alternative approach to case-control studies is to phenotype a general population sample for PI variants and compare the prevalence of COPD in PI MZ individuals with that in PI M individuals.34 Many of these studies are based on small numbers of individuals and have insufficient power to detect an effect of the PI MZ or PI MS phenotype. In a recent large cohort study the prevalence of obstructive pulmonary disease—defined as hospital discharge diagnosis of either asthma, chronic bronchitis or emphysema—was compared in 1551 PI MZ individuals and 14 484 controls from the general population of unknown PI genotype.36 The risk for obstructive pulmonary disease was significantly increased in the PI MZ individuals compared with the controls (relative risk (RR) = 2). However, only first degree relatives of PI Z COPD patients had an increased risk, suggesting that other genetic or environmental factors contribute to the increased risk in these patients. Most recently, Dahl et al37 have performed a large cross sectional study of 9187 individuals from the general population. Each individual underwent pulmonary function testing and was genotyped for the S and Z PI variants. Of the genotypic groups, only the PI SZ and PI ZZ individuals showed an increased prevalence of airflow obstruction (FEV1 <80% predicted). Neither the PI MS or PI MZ genotype was associated with a lower level of lung function in individuals without COPD. However, in patients with COPD FEV1 was 655 ml less in PI MZ individuals than in PI M individuals after adjustment for confounding variables. The observation that the PI MZ genotype was only associated with reduced lung function in those with COPD again suggests that other predisposing factors must exist.
3` PI mutation
Several polymorphisms of the PI gene are not associated with AAT deficiency—for example, a G→A transition in the 3` region of the gene. The A allele of this polymorphism was associated with COPD in some populations38,39 but not in others.40,41 The A allele was also associated with decreased transcription factor binding and decreased gene expression in vitro.42,43 The 3` mutation could therefore affect the acute phase response leading to attenuated upregulation of AAT synthesis when inflammation is present. In contrast to the in vitro data, the 3` polymorphism was not associated with a reduced AAT acute phase response in patients undergoing open heart surgery44 or in patients who had cystic fibrosis.45 The role of the 3` polymorphism in the pathogenesis of COPD therefore remains unclear.
α1-Antichymotrypsin
α1-Antichymotrypsin (AACT) is a protease inhibitor that is secreted by hepatocytes and alveolar macrophages. The gene contains several polymorphisms, and the Ala variant of an amino acid substitution (Pro227→Ala) that causes AACT deficiency was associated with COPD in one population46 but not others.41,47 This variant is rare and larger cohorts will be required to identify its role definitively in the pathogenesis of COPD. A recent Japanese study of 53 patients with COPD and 65 controls matched for age, sex, and smoking reported that homozygotes for the Ala variant of an amino acid substitution (Ala-15Thr) in the AACT signal peptide were increased in the patients compared with the controls (OR=3).48 This association is difficult to interpret since the Ala variant was not associated with AACT deficiency but it may reflect linkage disequilibrium with another allele.
Xenobiotic metabolising enzymes
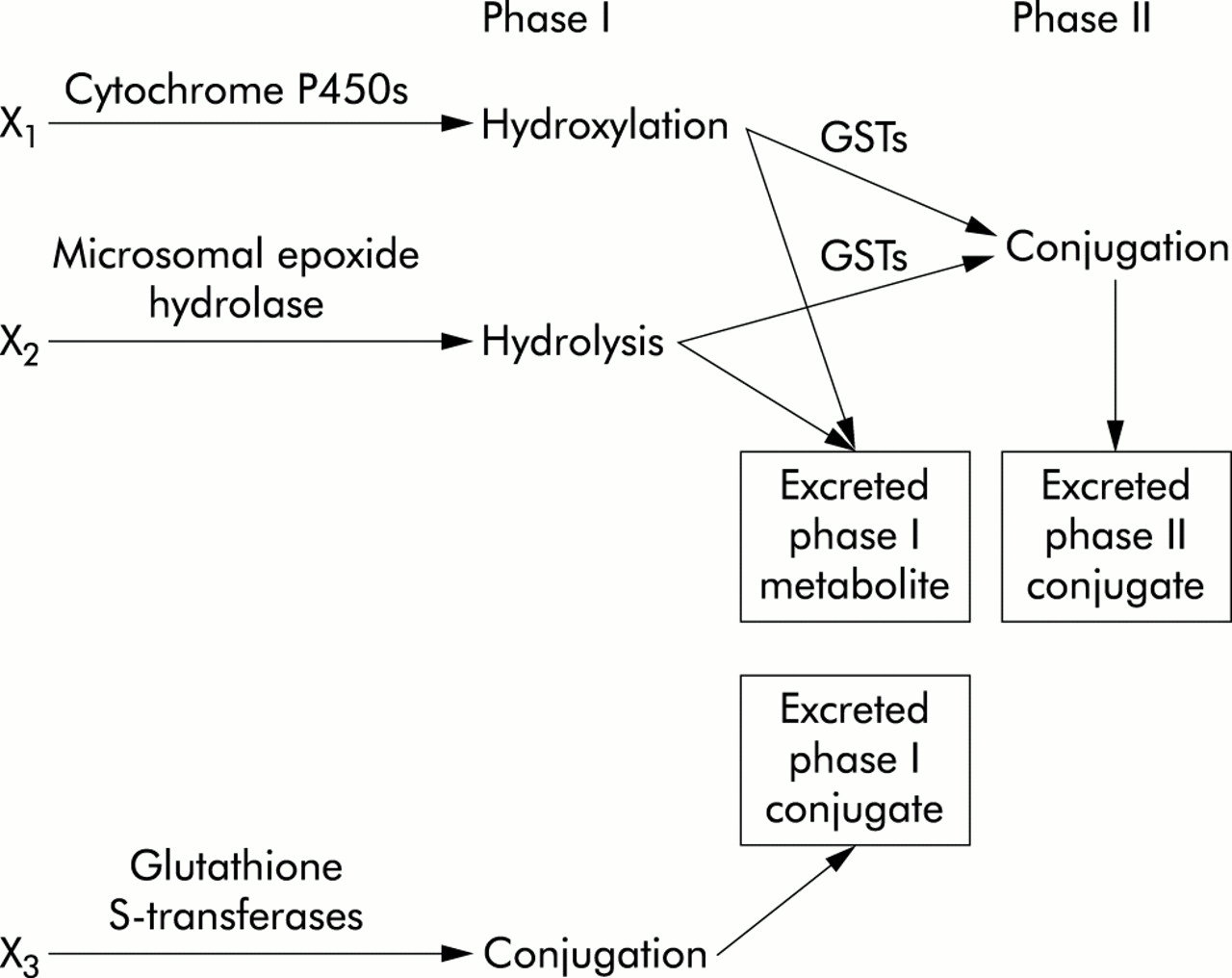
Figure 2 summarises the function of the xenobiotic metabolising enzymes that have been investigated as risk factors for the development of COPD.

{kind=link}
{kind=link}
Overview of xenobiotic metabolism focusing on genes investigated as candidates for COPD. X1, X2, and X3 are different xenobiotic substrates. GSTs = glutathione S-transferases.
Microsomal epoxide hydrolase
Microsomal epoxide hydrolase (EPHX1) is an enzyme that plays an important role in the metabolism of highly reactive epoxide intermediates formed in cigarette smoke. EPHX1 is expressed in several different cell types including hepatocytes and bronchial epithelial cells. Two common polymorphisms occur in the EPHX1 gene: Tyr113→His and His139→Arg. These polymorphisms correlated with the level of EPHX1 enzymatic activity in transfected cell lines49 but not in liver tissue samples.50 The slow metabolising form of EPHX1 was found in a higher proportion of patients with emphysema (22%) and COPD (19%) than in controls (6%), giving an odds ratio of 5.51 In a smaller Japanese study the slow metabolising form of EPHX1 was associated with more severe COPD.52 These results were not confirmed in a Korean population.53 However, EPHX1 genotypes were associated with rate of decline of lung function in smokers.54
Glutathione S-transferases
Glutathione S-transferases (GSTs) are enzymes that play an important role in detoxifying various aromatic hydrocarbons found in cigarette smoke. GSTM1 is expressed in the liver and the lung. Homozygous deletion of the GSTM1 gene occurs in approximately 50% of Caucasians and was associated with emphysema in patients who had lung cancer (OR=2)55 and with severe chronic bronchitis in heavy smokers (OR=3).56 However, in a Korean study there was no association of GSTM1 and GSTT1 polymorphisms with COPD.53
GSTP1 is expressed in the same cell types as GSTM1, although at a higher level.57 There is a polymorphism at position 105 (Ile105→Val), leading to an increased catalytic activity of the enzyme in vitro.58 Homozygotes for the isoleucine allele were significantly increased in Japanese patients with COPD compared with controls (OR=3).59
Antioxidants
Heme oxygenase-1
Heme oxygenase-1 (HMOX1) degrades heme to biliverdin and has been found to provide cellular protection against heme and non-heme mediated oxidant injury.60,61 A microsatellite (GT)n polymorphism within the heme oxygenase gene (HMOX1) promoter was associated with emphysema in Japanese smokers.62 In this study of 100 patients with emphysema and 101 matched controls, alleles with a high number of GT repeats (≥30) were more prevalent in the cases (21%) than in the controls (10%) yielding an odds ratio of 2. The authors hypothesised that the association was due to the effect of purine-pyrimidine repeats in promoting the formation of left handed Z DNA, thus downregulating gene expression.63 A high number of GT repeats in the HMOX1 promoter could suppress expression of the gene and leave the lung susceptible to oxidant injury. In support of this hypothesis, reporter gene constructs with low numbers of GT repeats upregulated gene expression in response to hydrogen peroxide whereas constructs with a higher number of repeats did not.62
Inflammatory mediators
Tumour necrosis factor α
Tumour necrosis factor α (TNFα) is a proinflammatory cytokine that has many effects relevant to the pathogenesis of COPD including neutrophil release from the bone marrow and neutrophil activation. The TNF gene contains a G→A transition in the promoter (TNF G-308A) that was associated with the level of TNF production in vitro.64 An association of the TNF -308A allele with COPD was found in a Taiwanese population (OR=11).65 The patients were selected based on the presence of chronic bronchitis and impaired lung function (FEV1 <80% predicted and FEV1/FVC <0.69). This association was confirmed recently in a Japanese study in which 106 patients with an FEV1 of <80% predicted and an FEV1/FVC of <0.7 were compared with 110 asymptomatic smokers/ex-smokers and 129 adult blood donors.66TNF -308A was significantly increased in the cases compared with both the control groups (OR=3). However, no association of the -308A allele with COPD was found in a study of 53 patients with COPD and 65 controls from a Japanese population.67 Studies of Caucasian populations have found no association of TNF -308A with COPD68 or rate of decline of lung function.54
Mucociliary clearance
Cystic fibrosis transmembrane regulator
In 1989 mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene were identified as the cause of cystic fibrosis (CF). CF carriers may also be predisposed to respiratory disease. IVS8 is a CFTR polymorphism consisting of a variable length thymine repeat in intron 8 of the gene. The IVS8-5T allele results in reduced CFTR gene expression. Studies of IVS8-5T as a risk factor for COPD have yielded conflicting results.69,70 A recent study by Tzetis et al71 in a Greek population of 12 patients with obstructive lung diseases and 52 controls screened for variants in the entire CFTR coding region found no significant increase in CF-causing mutations in the patients compared with the controls. The frequency of the Met allele of the Met470Val polymorphism was increased in the patients (71%) compared with the controls (36%). However, the Met470 variant is associated with increased CFTR chloride channel activity72 and therefore the reason for the association with COPD is unclear.
Other genes
Several other genes have been investigated as risk factors for the development of COPD and these are listed in table 1.
Genes investigated as candidates for COPD
FUTURE STUDIES OF GENOTYPE–ENVIRONMENT INTERACTION
In most of the studies discussed in this review there has been little specific consideration of genotype–environment interactions. Of interest, segregation analysis and linkage analysis typically assume that no genotype–environment interaction is present. Approaches which include genotype–environment interactions in segregation analysis have been developed, but they have not been applied in COPD.73 New methods of including genotype–environment interaction in linkage analysis and family based association analysis have also been developed, which may be useful in future studies of the genetics of COPD.73,74 In addition to assessing the significance of genotype–environment interactions, inclusion of these interactions in linkage and association studies may actually improve the detection of main genetic effects because the relationship between genetic and environmental factors is modelled more accurately.
The most commonly performed design used in the study of genetics in COPD has been the case-control genetic association study. Future association studies may also be strengthened by the inclusion of environmental risk factors in the study design and analysis. The methodology employed could be a case-control approach using multivariate regression models to examine interactions. An alternative methodology could be to use a case only design75 if the genotype and environmental risk factors are independent.76
In conclusion, genotype–environment interactions are likely to be essential contributors to the development of COPD. Intensified efforts to identify these interactions may lead to improved understanding of the pathogenesis of the disease.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 31a.↵
- 31b.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.
- 81.
- 82.↵
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.
- 91.↵