Article Text

Abstract
Colonisation with Pseudomonas aeruginosa is common in adults with cystic fibrosis (CF) and there is increasing evidence that transmissible strains may cross colonise patients. However, transmission of these strains by social contact to healthy non-CF individuals has not been described. A case is presented where an adult CF patient colonised by an epidemic P aeruginosa strain infected her parents with subsequent morbidity.
- Pseudomonas aeruginosa
- cystic fibrosis
- cross infection
Statistics from Altmetric.com
P seudomonas aeruginosa is a well recognised pathogen in patients with cystic fibrosis (CF) and up to 80% of the adult population are chronically colonised.1 While it was assumed that patients were infected by individual unique strains, with shared strains only occurring between CF siblings,2 there is now increasing evidence that many patients with CF may be colonised by the same (epidemic) strains, with transmission occurring between unrelated CF patients.3,4
However, healthy non-CF relatives of these patients are not thought to be at risk from cross infection by P aeruginosa as this organism is not described as a primary pathogen in non-CF lungs. We present a case in which a patient with CF chronically colonised with an epidemic P aeruginosa strain cross infected her non-CF parents with subsequent significant morbidity.
We subsequently looked for P aeruginosa carriage in the relatives of a group of our CF patients colonised by this epidemic strain.
CASE REPORT
Index case
The index case (age 22 years, DF508/G551D) was diagnosed with CF at birth due to meconium ileus. She became chronically colonised with P aeruginosa in 1981 while attending the local paediatric hospital. In 1995 genotyping of her P aeruginosa isolates (by pulsed field gel electrophoresis (PFGE)) revealed that she was colonised by a multiresistant epidemic P aeruginosa strain which was prevalent in the paediatric unit at the time.3 She is currently stable with a good nutritional state (BMI 22 kg/m2) and spirometric status (FEV1 95% predicted), and usually requires three courses of intravenous antibiotics per year. She lives with her parents who assist with chest physiotherapy twice daily. When well she produces small amounts of sputum only. Regular genotypic analysis of her sputum cultures reveals that she is chronically colonised by two different P aeruginosa strains, the epidemic strain and a second unique β-lactam resistant strain.
Infection of the mother
In February 2000 her mother aged 56 years presented to her GP with a 5 month history of flu-like symptoms and pleuritic chest pain. She had no past illnesses of note apart from mild asthma and was previously well. She had lost 6 kg in 2 months and was producing 30 ml sputum/day. A sputum sample was sent for microbiological analysis. She was commenced on amoxycillin 250 mg tds for 7 days and ciprofloxacin 500 mg bd for 7 days by her GP with no clinical improvement. Sputum samples grew fully sensitive Streptococcus pneumoniae and a P aeruginosa strain resistant to ceftazidime (MIC >32), and she was referred to our unit.
At presentation she was pyrexial with bilateral chest wheezes and a clear chest radiograph. FEV1 was 83% predicted. Microbiological samples sent for tuberculous culture were negative, as were serological tests for mycoplasma, legionella, and respiratory viruses. A sputum sample taken on admission cultured only a ceftazidime resistant strain of P aeruginosa. She was admitted for intravenous antibiotics (piperacillin 4 g tds and ciprofloxacin 400 mg bd) and discharged with normal spirometric parameters after 1 week. Sputum culture was negative 1 month later but her symptoms subsequently returned and multiresistant P aeruginosa was re-isolated and has been present in all subsequent sputum samples, despite repeated attempts at eradication. A high resolution CT lung scan revealed no evidence of bronchiectasis. Sweat testing produced a low normal result. Genetic testing for CF revealed her to be a DF508 carrier only. Extensive second level screening (ruling out 99% of CF genes) failed to reveal a second CF gene.
Infection of the father
In March 2000 her father aged 64 years was transferred from a local hospital where he had been admitted with pneumonia. He had a past history of right upper lobectomy and thoracoplasty in 1971 for a bronchiectatic illness, and was being treated for mild obstructive airways disease with inhaled medication. There was no history of tuberculosis and previous sputum cultures had never grown Pseudomonas sp.
On admission he was unwell, pyrexial, and had a pleural rub over the lingula. Chest radiography showed the right thoracoplasty and new lingula consolidation (fig 1). Sputum analysis revealed two P aeruginosa phenotypes with identical ceftazidime resistant antibiograms. No other organisms were isolated. He was commenced on intravenous colomycin and tobramycin and made a slow recovery. On discharge from hospital after 3 weeks the lingula consolidation had cleared and repeat sputum culture was negative. Sputum cultures have subsequently repeatedly grown a ceftazidime resistant P aeruginosa strain, indicating chronic colonisation. A subsequent high resolution CT scan of the chest revealed minor bronchiectatic changes in the residual right lung. Genetic testing for CF genes revealed him to be a G551D carrier only.

Chest radiograph of the father taken on admission showing right thoracoplasty and new lingula consolidation.
Genotyping
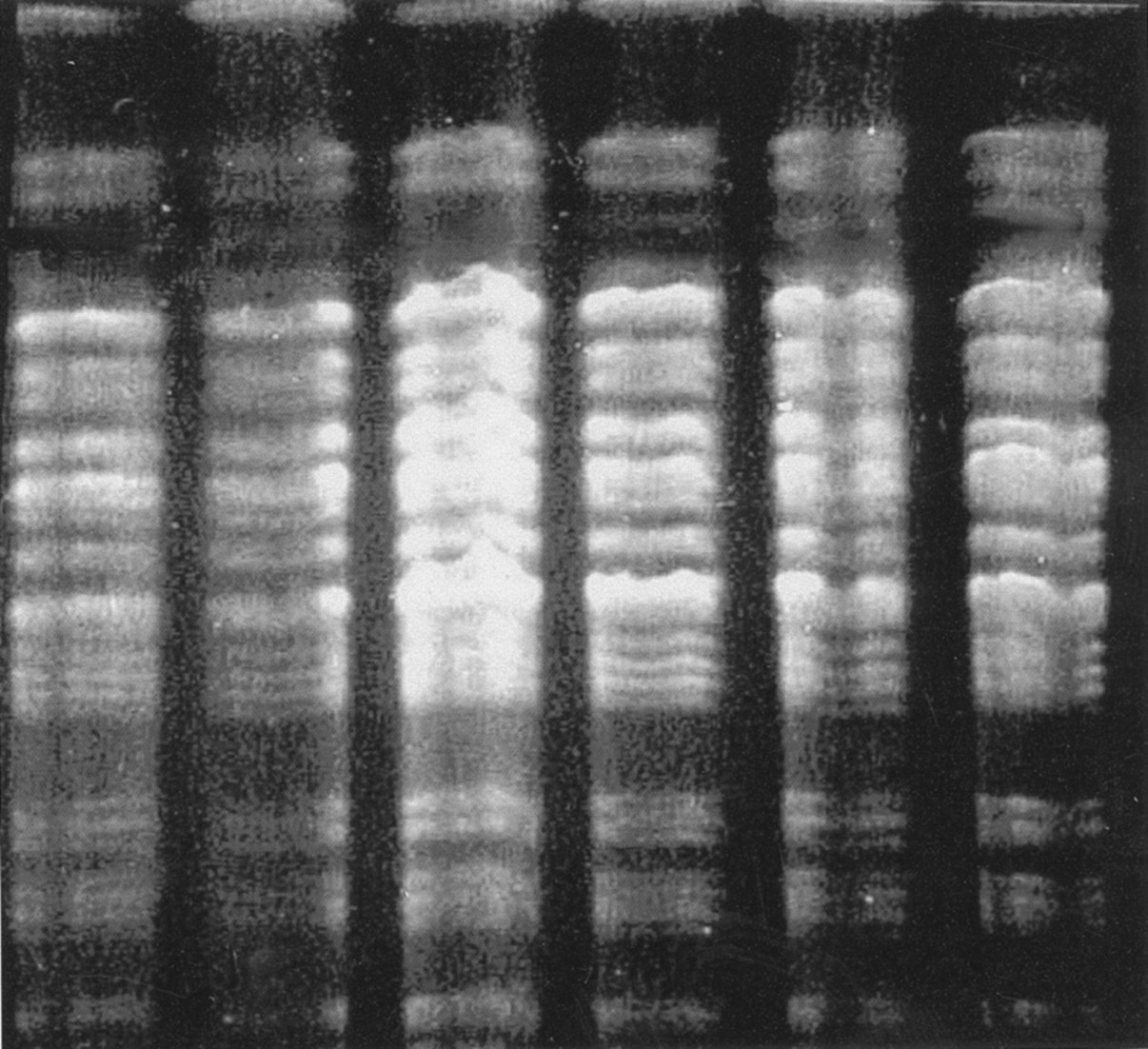
Chromosomal DNA of each P aeruginosa isolate from sputum samples of the patient and her parents was genotypically analysed by PFGE using pulses of 5–33 at 6 V/cm for 20 hours following digestion with the endonuclease Spe1. Using the criteria of Tenover et al,5 this revealed that they were identical to each other and also to the original stored isolate from 1995, confirming that all three individuals were colonised by the same epidemic strain (fig 2).
{kind=link}
{kind=link}
Pulsed field gel electrophoresis in 1% pulse field agarose after cutting with Spe 1. Lane 1, reference AH strain; lane 2, index CF patient's stored strain; lane 3, index CF patient's current strain; lane 4, mother's initial strain; lane 5, mother's subsequent strain; lane 6, father's strain. All genotypes are identical, confirming Pseudomonas aeruginosa strain AH.
Study of relatives
We subsequently analysed gargle samples from the throats of 31 relatives (12 mothers, six fathers, two grandparents, one sibling, and 10 partners) of 23 patients with CF colonised by this epidemic strain. All individuals had no chest symptoms. They gargled with 5 ml sterile water for 1 minute and the resulting samples were immediately stored at –70°C for later batched analysis. The thawed samples were spun and the cell debris plated onto blood agar plates and incubated at 37°C for 48 hours. Although P aeruginosa was cultured from two of these samples, PFGE analysis of the isolates demonstrated that they were unique and therefore presumably environmental.
DISCUSSION
It is well recognised that patients with CF are susceptible to infection and subsequent colonisation with P aeruginosa due to the abnormal environment found in the CF lung. Most patients are colonised by unique strains, but in 1996 Cheng et al reported that 85% of children with CF at our local paediatric hospital colonised by P aeruginosa had the same epidemic strain.3 They proposed that this strain was able to cross infect patients with CF and advocated a policy of strict segregation to prevent further cases. While infection with P aeruginosa in non-CF individuals is well described, it only occurs in those with preceding bronchiectasis, the immunocompromised, or those undergoing mechanical ventilation. P aeruginosa is not recognised as a primary respiratory pathogen in otherwise healthy adult non-CF individuals.
We have demonstrated infection by P aeruginosa and subsequent chronic colonisation in the healthy parents of a patient with CF who must have been cross infected by their colonised daughter. Why these individuals became cross infected remains unclear. Cross infection only occurred with the epidemic P aeruginosa strain, which confirms that strains vary in their transmissibility, and we have recently shown that this strain can superinfect CF patients who are already colonised by unique Pseudomonas strains.6 However, it is unclear why the parents remain colonised. Neither parent habitually uses a nebuliser, which could be a source of recurrent infection,7 nor do they have symptoms of sinus colonisation. While the father had pre-existing lung disease, the mother had no evidence of gross bronchiectasis on the CT scan. We postulate that both parents must have a degree of bronchial wall damage to allow persistent colonisation and, because they live in close contact with their chronically colonised CF daughter, it is possible that she is continually re-infecting them.
This is a worrying development since parents are intimately involved in the treatment schedules of young patients with CF who rely upon them to supervise chest physiotherapy. Furthermore, this strain is multiresistant and difficult to treat. In both our cases cross infection produced serious morbidity requiring admission to hospital, and both parents have become recolonised despite initial eradication with recrudescence of symptoms in the mother. This has implications for the treatment of other non-CF family members who develop chest symptoms and the potential risk to hospital staff who are chronically exposed to this patient group.
It is reassuring that our study of other relatives and partners of patients with CF chronically colonised with this epidemic strain has failed to reveal colonisation with the organism. Nevertheless, we suggest that individuals continually exposed to CF patients colonised with transmissible strains (including hospital staff) who develop persistent chest symptoms be screened for this organism.