Article Text

Abstract
Background: Idiopathic pulmonary alveolar proteinosis (PAP) has recently been recognised as a disease of impaired alveolar macrophage function caused by neutralising anti-granulocyte-macrophage colony-stimulating (anti-GM-CSF) autoantibodies. Subcutaneous recombinant human GM-CSF is a novel treatment for PAP, but its mechanism of action is unclear.
Methods: Clinical, functional, and bronchoalveolar lavage (BAL) findings were prospectively evaluated in a patient with PAP treated with daily subcutaneous GM-CSF 8 μg/kg for 12 weeks.
Results: Treatment resulted in improvements in dyspnoea, lung function, and peak cycle ergometry performance. In serum and BAL fluid the titre of anti-GM-CSF autoantibodies was raised at baseline and markedly reduced on treatment. At baseline the BAL fluid cellular profile showed a decrease in the absolute number and the percentage of macrophages (50%) and an increase in lymphocytes (45%), predominantly CD4+. This cellular distribution remained unchanged after 6 and 12 weeks of treatment while macrophages became morphologically normal and functionally improved. Extracellular proteinaceous material completely disappeared.
Conclusions: Clinically successful treatment of PAP with GM-CSF was associated with a profound reduction in GM-CSF neutralising autoantibodies, improvement in alveolar macrophage morphology and function, but persistent BAL lymphocytosis.
- pulmonary alveolar proteinosis
- bronchoalveolar lavage
- alveolar macrophages
- lymphocytic alveolitis
- granulocyte-macrophage colony-stimulating factor
Statistics from Altmetric.com
- pulmonary alveolar proteinosis
- bronchoalveolar lavage
- alveolar macrophages
- lymphocytic alveolitis
- granulocyte-macrophage colony-stimulating factor
Clinical aspects and current concepts on the pathogenesis of idiopathic pulmonary alveolar proteinosis (PAP) have recently been reviewed in detail.1 PAP is characterised by excessive intra-alveolar accumulation of surfactant phospholipids and protein caused by impaired macrophage function. The detection of a neutralising IgG targeting GM-CSF (anti-GM-CSF antibody) which was found in all tested cases of idiopathic PAP provides insights into the likely pathophysiological process.2 Treatment with subcutaneous recombinant human GM-CSF (rhGM-CSF) was successful in 43% of patients with PAP,3 but the mode of action of GM-CSF remains unclear.
In the present case study the effects of treatment with GM-CSF on cellular bronchoalveolar lavage (BAL) profiles, on the morphological characteristics of alveolar macrophages, and on the GM-CSF autoantibody titres in serum and BAL fluid were prospectively investigated.
PATIENT AND METHODS
A 40 year old woman had presented 6 years previously with progressive dyspnoea and extensive pulmonary infiltrates. PAP was diagnosed histopathologically on video-assisted thoracoscopic lung biopsy. Three recurrent severe restrictive and hypoxaemic episodes were treated with two sets of repeated whole lung lavages and one successful 12 week course of subcutaneous GM-CSF. One year later her condition deteriorated and, having given informed consent, she was included in the current study which was approved by the local and national ethics committees.
Bacterially synthesised rhGM-CSF 8 μg/kg (Leucomax; Essex Chemie AG, Lucerne, Switzerland) was administered subcutaneously once daily.3 Lung function tests and gas exchange, spiroergometry on a cycle ergometer, and bronchoscopy with BAL were performed before treatment and after 6 and 12 weeks.
Macrophage morphology
BAL fluid was centrifuged and the protein content of the supernatant was measured chemically. The cells were counted in a haemocytometer (Coulter counter T540, Instrumentation Laboratory Inc). A manual differential count of 400 Giemsa stained cells was performed on cytocentrifuge slides. Expression of the lymphocyte cell surface antigens CD3, CD4, and CD8 was measured by flow cytometry (Coulter-epics counter). Alveolar macrophages were classified into three morphological groups adapted from the classification of Iyonaga et al.4: (a) small monocyte-like cells without visible cytoplasmic accumulation of proteinaceous material, (b) foamy cells with visible cytoplasmic accumulation of proteinaceous material, and (c) huge ghost-like macrophages.
Macrophage adhesion test
After centrifugation and cell counting, BAL cells were resuspended in RPMI 1640 with 0.5% bovine serum albumin and placed in flat bottom wells (Costar) at a concentration of 2 × 106 cells/well. The number of macrophages per well was calculated according to the percentage in the Giemsa stained slides. Adhesion was allowed by incubation at 37°C. After 2 hours the suspension with the non-adherent cells was collected from the wells and the concentration of cells in the pooled suspension was again counted by flow cytometry (Coulter) and the percentage of macrophages determined in a Giemsa stained slide. The number of macrophages still present in the suspension was calculated and subtracted from the initial number of macrophages, assuming that all macrophages lost were adherent.
Anti-GM-CSF antibodies
Anti-GM-CSF antibodies in serum and in BAL fluid were assayed centrally in a blind manner. The antibody was purified by 33% ammonium sulphate sedimentation followed by affinity chromatography using NHS activated HiTrap (Amersham Pharmacia Biotech UK Ltd) previously coupled with rhGM-CSF (Escherichia coli derived; Kirin Brewery Co Ltd, Gunma, Japan). The purity of the isolated antibody was confirmed by SDS gel electrophoresis. The concentration of purified antibody was determined by a sandwich ELISA with non-labelled and peroxidase labelled anti-human IgG antibodies (Dako Corporation, Carpinteria, CA, USA) using human IgG as a standard for quantitative analysis. The titration of antibody was performed according to the method previously described5 with slight modification. Briefly, serum samples were diluted 1500 fold with phosphate buffered saline (PBS) containing 0.1% goat serum and 0.1% Tween 20. Fifty μl of diluted serum and the purified antibody as a standard were transferred to a plate coated with 1 μg/ml rhGM-CSF overnight at 4°C and blocked with 1% bovine serum albumin/PBS for 1 hour at room temperature. After washing four times with PBS 0.1% Tween 20, 50 μl of 10 mM ammonium acetate buffer (pH 5.0) was transferred into each well and kept at room temperature for 15 minutes, then each well was washed with PBS. The antibodies captured by rhGM-CSF were detected by peroxidase labelled anti-human IgG F(ab)2 antibody (Dako Corporation). After washing, colour was developed using tetramethylbenzidine and absorbance was measured at 450 nm. The concentration of antibody was measured from the standard curve.
RESULTS
Administration of subcutaneous GM-CSF was well tolerated and no side effects occurred. At the end of the treatment all pulmonary symptoms had disappeared. Pulmonary function, gas exchange at rest, and peak cycle ergometry performance were normal (table 1).
Clinical and bronchoalveolar lavage findings before and during a 12 week treatment course with daily subcutaneous granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment of idiopathic pulmonary alveolar proteinosis
The bronchoscopic examinations were performed without complications. The BAL fluid appeared opaque at baseline and cleared after 6 and 12 weeks, reflected by a decrease in the total protein content of the supernatant from 2.0 g/l to 0.2 g/l. BAL cell counts and cellular profiles decreased in both absolute numbers and in the percentage of macrophages (300 cells/μl, 50%) and lymphocytes increased (276 cells/μl, 46%) with a 75% predominance of CD4+ lymphocytes (CD4/CD8 ratio 3.7) which remained unchanged during treatment (table 1). Microscopic examination at baseline showed frequent cell fragments (fig 1A) and large masses of mostly extracellular amorphous PAS-positive material (fig 1B). Morphologically, most macrophages appeared as “ghost cells” with fewer large foamy macrophages. After 6 weeks of GM-CSF treatment (fig 1C and D) cell debris and fragments had almost completely disappeared and extracellular amorphous material was no longer detectable. The macrophage population consisted of small monocyte-like cells without intracellular proteinaceous material (33%), larger foamy macrophages with intracellular proteinaceous material (62%), and a few ghost-like huge macrophages (3%). After 12 weeks of treatment with GM-CSF (fig 1E and F) the macrophage population again changed their appearance: their morphology was homogenous with 90% foamy protein loaded macrophages, only 10% of small monocyte-like macrophages, and ghost-like cells were no longer detectable.
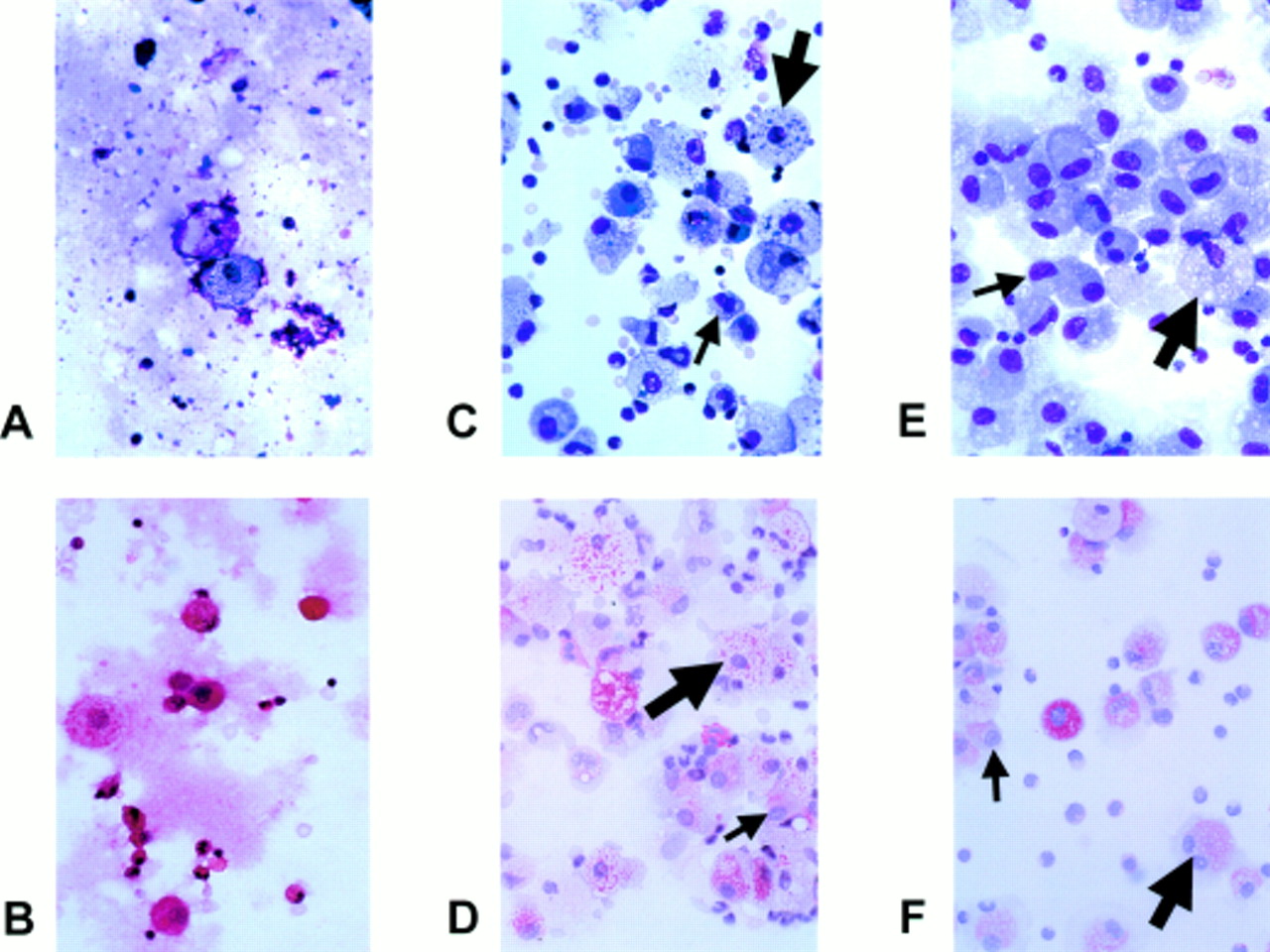
{kind=link}
Bronchoalveolar lavage findings of a patient with pulmonary alveolar proteinosis before (A and B), after 6 weeks (C and D), and after 12 weeks (E and F) of treatment with subcutaneous GM-CSF. Note the cloudy intracellular and extracellular proteinaceous material and the damaged alveolar macrophages with cell fragments before treatment in A and B. After 6 weeks of treatment with GM-CSF (C and D) cell debris and extracellular proteinaceous material have almost completely disappeared. Macrophages are morphologically normal and consist of small monocyte-like cells (small arrow) and larger foamy macrophages with ingested proteinaceous material (large arrow). After 12 weeks of treatment (E and F) the macrophages mainly consist of foamy macrophages (large arrow) with a few small monocyte-like cells (small arrow). BAL lymphocytosis is present before and during treatment with GM-CSF. A, C, E: Giemsa stain; B, D, F: PAS stain.
Macrophage adhesion to tissue culture plastic was low before treatment (75%) but, after 6 and 12 weeks of treatment, adhesion increased (95%).
The titre of anti-GM-CSF antibody in both BAL fluid and serum declined throughout treatment. The effect was remarkable in BAL fluid with a reduction from 4.4 μg/ml before treatment to 0.14 μg/ml after 6 weeks of treatment with GM-CSF and 0.13 μg/ml at 12 weeks. In serum the titre fell from 35.2 μg/ml at day 12 to 20.6 μg/ml after treatment (table 1).
DISCUSSION
PAP is currently recognised as a disease of impaired alveolar macrophage function leading to excessive accumulation of surfactant components in the alveoli.1 To our knowledge, this is the first study to investigate BAL findings during successful GM-CSF treatment for human PAP. The observed changes in the morphology and function of alveolar macrophages over time were profound. Before treatment, cell fragments and huge ghost-like macrophages dominated and extracellular proteinaceous material was abundant. During treatment with GM-CSF, younger monocyte-like macrophages appeared, the huge macrophages with the cell fragments disappeared, and the extracellular proteinaceous material was no longer detectable.
Alveolar macrophages in PAP show less chemotactic and phagocytic activity and reduced cellular adherence.1 Storage of excess surfactant may lead to macrophage ghost cells with impaired function.1 In our patient GM-CSF was able to improve macrophage morphology and function, as shown in vivo by the clearance of surfactant proteins and in vitro by increased adherence to tissue culture plastic. The reduction in the GM-CSF neutralising antibody titre in serum and BAL fluid suggests that neutralisation of GM-CSF activity may be responsible for the impaired macrophage function. GM-CSF may induce proliferation and maturation of progenitor cells in the bone marrow,6 recruit additional cells from the blood, and promote the proliferation of alveolar macrophages present in the lung, an effect which has been shown in vitro.7 GM-CSF is also able to augment phagocytosis, as shown in other diseases such as mycobacterial infection8 and in cancer to clear apoptotic cells.9
The presence of anti-GM-CSF antibody classifies PAP as an autoimmune disease. In this respect, treatment with GM-CSF raises concern because it may actually induce autoantibodies.10 However, in our patient the titre of autoantibodies in BAL fluid and in the serum was gradually lowered during treatment with GM-CSF.
BAL lymphocytosis in primary PAP, with a predominance of CD4+ cells in most cases, has been reported previously.11 In our patient BAL lymphocytosis persisted throughout the treatment course, despite clinical improvement and disappearance of surfactant from the alveolar space. The hypothesis that BAL lymphocytosis may be a consequence of surfactant accumulation in the lung11 is thus not supported by our observation. Interestingly, lymphocytic perivascular and peribronchiolar infiltrates were also found in the animal models of PAP.12 Under successful treatment with aerosolised GM-CSF, these lymphocytic infiltrates were less pronounced but remained present.13 The pathogenetic role of this persistent lymphocytosis remains to be determined.
In conclusion, successful GM-CSF treatment of PAP was associated with an improvement in alveolar macrophage morphology and function, a profound reduction in anti-GM-CSF antibodies, but persistent BAL lymphocytosis.
Acknowledgments
The authors thank R Rüegg, chief technician, Hematology Laboratory, University Hospital Zurich, Switzerland, for her expertise and help in evaluating the BAL cell morphology, Dr N Keicho and Dr K Uchida for analysing the anti-GM-CSF antibodies, and Essex Chemie AG (Lucerne, Switzerland) for the provision of recombinant human GM-CSF.
REFERENCES
Footnotes
-
Funding: Silva Casa Foundation Switzerland, Swiss Respiratory Society, Swiss National Science Foundation.