Article Text

Abstract
Early detection and correction of tissue hypoxia is essential if progressive organ dysfunction and death are to be avoided. However, hypoxia in individual tissues or organs caused by disordered regional distribution of oxygen delivery or disruption of the processes of cellular oxygen uptake and utilisation cannot be identified from global measurements. Regional oxygen transport and cellular utilisation have an important role in maintaining tissue function. When tissue hypoxia is recognised, treatment must be aimed at the primary cause. Supplemental oxygen may be life saving in some situations but cannot correct inadequate oxygen delivery caused by a low cardiac output or impaired ventilation. Recent innovations include artificial oxygen carrying proteins and “haemoglobin” molecules designed to improve tissue blood flow by reducing viscosity. Regulating cell metabolism using different substrates or drugs has so far been poorly explored but is an exciting area for further research. A minimum level of global oxygen delivery and perfusion pressure must be maintained in the critically ill patient with established “shock”, but advances in the understanding and control of regional distribution and other “downstream” factors in the oxygen cascade are needed to improve outcome in these patients.
- intensive care
- oxygen delivery
- So2, oxygen saturation (%)
- Po2, oxygen partial pressure (kPa)
- Pio2inspired Po2
- Peo2mixed expired Po2
- Peco2mixed expired Pco2
- Pao2alveolar Po2
- Pao2arterial Po2
- Sao2arterial So2
- Svo2mixed venous So2
- Qt, cardiac output
- Hb, haemoglobin
- Cao2, arterial O2 content
- Cvo2, mixed venous O2 content
- Vo2, oxygen consumption
- Vco2, CO2 production
- O2R, oxygen return
- Do2, oxygen delivery
- Vi/e, minute volume, inspiratory/expiratory
Statistics from Altmetric.com
- So2, oxygen saturation (%)
- Po2, oxygen partial pressure (kPa)
- Pio2inspired Po2
- Peo2mixed expired Po2
- Peco2mixed expired Pco2
- Pao2alveolar Po2
- Pao2arterial Po2
- Sao2arterial So2
- Svo2mixed venous So2
- Qt, cardiac output
- Hb, haemoglobin
- Cao2, arterial O2 content
- Cvo2, mixed venous O2 content
- Vo2, oxygen consumption
- Vco2, CO2 production
- O2R, oxygen return
- Do2, oxygen delivery
- Vi/e, minute volume, inspiratory/expiratory
Although traditionally interested in conditions affecting gas exchange within the lungs, the respiratory physician is increasingly, and appropriately, involved in the care of critically ill patients and therefore should be concerned with systemic as well as pulmonary oxygen transport. Oxygen is the substrate that cells use in the greatest quantity and upon which aerobic metabolism and cell integrity depend. Since the tissues have no storage system for oxygen, a continuous supply at a rate that matches changing metabolic requirements is necessary to maintain aerobic metabolism and normal cellular function. Failure of oxygen supply to meet metabolic needs is the feature common to all forms of circulatory failure or “shock”. Prevention, early identification, and correction of tissue hypoxia are therefore necessary skills in managing the critically ill patient and this requires an understanding of oxygen transport, delivery, and consumption.
OXYGEN TRANSPORT
Oxygen transport describes the process by which oxygen from the atmosphere is supplied to the tissues as shown in fig 1⇓ in which typical values are quoted for a healthy 75 kg individual. The phases in this process are either convective or diffusive: (1) the convective or “bulk flow” phases are alveolar ventilation and transport in the blood from the pulmonary to the systemic microcirculation: these are energy requiring stages that rely on work performed by the respiratory and cardiac “pumps”; and (2) the diffusive phases are the movement of oxygen from alveolus to pulmonary capillary and from systemic capillary to cell: these stages are passive and depend on the gradient of oxygen partial pressures, the tissue capillary density (which determines diffusion distance), and the ability of the cell to take up and use oxygen.

Oxygen transport from atmosphere to mitochondria. Values in parentheses for a normal 75 kg individual (BSA 1.7 m2) breathing air (Fio2 0.21) at standard atmospheric pressure (PB 101 kPa). Partial pressures of O2 and CO2 (Po2, Pco2) in kPa; saturation in %; contents (Cao2, Cvo2) in ml/l; Hb in g/l; blood/gas flows (Qt, Vi/e) in l/min. P50 = position of oxygen haemoglobin dissociation curve; it is Po2 at which 50% of haemoglobin is saturated (normally 3.5 kPa). Do2 = oxygen delivery; Vo2 = oxygen consumption, Vco2 = carbon dioxide production; Pio2, Peo2 = inspired and mixed expired Po2; Peco2 = mixed expired Pco2; Pao2 = alveolar Po2.
This review will not consider oxygen transport within the lungs but will focus on transport from the heart to non-pulmonary tissues, dealing specifically with global and regional oxygen delivery, the relationship between oxygen delivery and consumption, and some of the recent evidence relating to the uptake and utilisation of oxygen at the tissue and cellular level.
OXYGEN DELIVERY
Global oxygen delivery (Do2) is the total amount of oxygen delivered to the tissues per minute irrespective of the distribution of blood flow. Under resting conditions with normal distribution of cardiac output it is more than adequate to meet the total oxygen requirements of the tissues (Vo2) and ensure that aerobic metabolism is maintained.
Recognition of inadequate global Do2 can be difficult in the early stages because the clinical features are often non-specific. Progressive metabolic acidosis, hyperlactataemia, and falling mixed venous oxygen saturation (Svo2), as well as organ specific features such as oliguria and impaired level of consciousness, suggest inadequate Do2. Serial lactate measurements can indicate both progression of the underlying problem and the response to treatment. Raised lactate levels (>2 mmol/l) may be caused by either increased production or reduced hepatic metabolism. Both mechanisms frequently apply in the critically ill patient since a marked reduction in Do2 produces global tissue ischaemia and impairs liver function.
Table 1⇓ illustrates the calculation of Do2 from the oxygen content of arterial blood (Cao2) and cardiac output (Qt) with examples for a normal subject and a patient presenting with hypoxaemia, anaemia, and a reduced Qt. The effects of providing an increased inspired oxygen concentration, red blood cell transfusion, and increasing cardiac output are shown. This emphasises that: (1) Do2 may be compromised by anaemia, oxygen desaturation, and a low cardiac output, either singly or in combination; (2) global Do2 depends on oxygen saturation rather than partial pressure and there is therefore little extra benefit in increasing Pao2 above 9 kPa since, due to the sigmoid shape of the oxyhaemoglobin dissociation curve, over 90% of haemoglobin (Hb) is already saturated with oxygen at that level. This does not apply to the diffusive component of oxygen transport that does depend on the gradient of oxygen partial pressure.
Relative effects of changes in Pao2, haemoglobin (Hb), and cardiac output (Qt) on oxygen delivery (Do2)
Although blood transfusion to polycythaemic levels might seem an appropriate way to increase Do2, blood viscosity increases markedly above 100 g/l. This impairs flow and oxygen delivery, particularly in smaller vessels and when the perfusion pressure is reduced, and will therefore exacerbate tissue hypoxia.1 Recent evidence suggests that even the traditionally accepted Hb concentration for critically ill patients of approximately 100 g/l may be too high since an improved outcome was observed if Hb was maintained between 70 and 90 g/l with the exception of patients with coronary artery disease in whom a level of 100 g/l remains appropriate.2 With the appropriate Hb achieved by transfusion, and since the oxygen saturation (Sao2) can usually be maintained above 90% with supplemental oxygen (or if necessary by intubation and mechanical ventilation), cardiac output is the variable that is most often manipulated to achieve the desired global Do2 levels.
OXYGEN CONSUMPTION
Global oxygen consumption (Vo2) measures the total amount of oxygen consumed by the tissues per minute. It can be measured directly from inspired and mixed expired oxygen concentrations and expired minute volume, or derived from the cardiac output (Qt) and arterial and venous oxygen contents:
Vo2 = Qt × (Cao2 – Cvo2)
Directly measured Vo2 is slightly greater than the derived value that does not include alveolar oxygen consumption. It is important to use the directly measured rather than the derived value when studying the relationship between Vo2 and Do2 to avoid problems of mathematical linkage.3
The amount of oxygen consumed (Vo2) as a fraction of oxygen delivery (Do2) defines the oxygen extraction ratio (OER):
OER = Vo2/Do2
In a normal 75 kg adult undertaking routine activities, Vo2 is approximately 250 ml/min with an OER of 25% (fig 1⇑), which increases to 70–80% during maximal exercise in the well trained athlete. The oxygen not extracted by the tissues returns to the lungs and the mixed venous saturation (Svo2) measured in the pulmonary artery represents the pooled venous saturation from all organs. It is influenced by changes in both global Do2 and Vo2 and, provided the microcirculation and the mechanisms for cellular oxygen uptake are intact, a value above 70% indicates that global Do2 is adequate.
A mixed venous sample is necessary because the saturation of venous blood from different organs varies considerably. For example, the hepatic venous saturation is usually 40–50% but the renal venous saturation may exceed 80%, reflecting the considerable difference in the balance between the metabolic requirements of these organs and their individual oxygen deliveries.
CLINICAL FACTORS AFFECTING METABOLIC RATE AND OXYGEN CONSUMPTION
The cellular metabolic rate determines Vo2. The metabolic rate increases during physical activity, with shivering, hyperthermia and raised sympathetic drive (pain, anxiety). Similarly, certain drugs such as adrenaline4 and feeding regimens containing excessive glucose increase Vo2. Mechanical ventilation eliminates the metabolic cost of breathing which, although normally less than 5% of the total Vo2, may rise to 30% in the catabolic critically ill patient with respiratory distress. It allows the patient to be sedated, given analgesia and, if necessary, paralysed, further reducing Vo2.
RELATIONSHIP BETWEEN OXYGEN CONSUMPTION AND DELIVERY
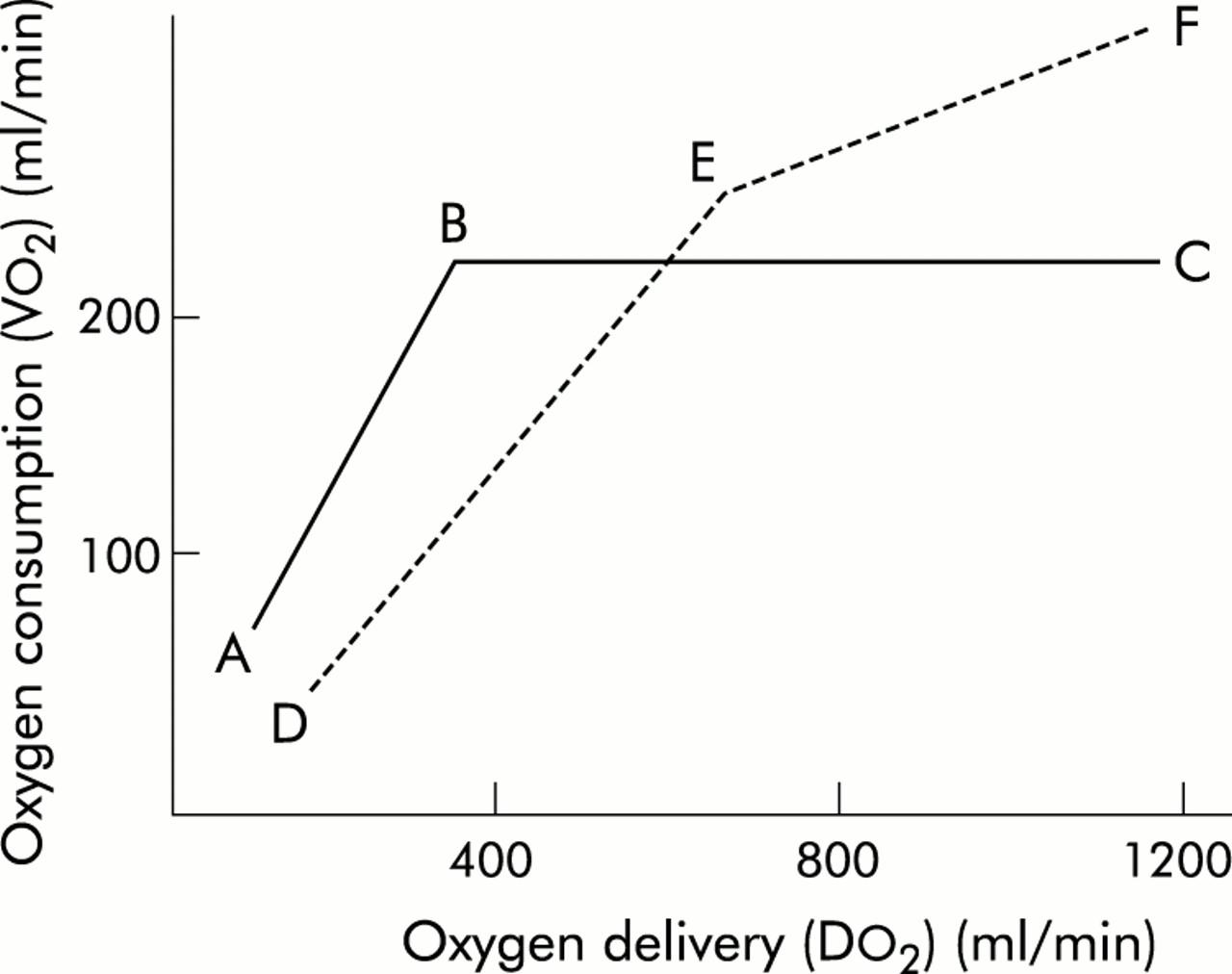
The normal relationship between Vo2 and Do2 is illustrated by line ABC in fig 2⇓. As metabolic demand (Vo2) increases or Do2 diminishes (C–B), OER rises to maintain aerobic metabolism and consumption remains independent of delivery. However, at point B—called critical Do2 (cDo2)—the maximum OER is reached. This is believed to be 60–70% and beyond this point any further increase in Vo2 or decline in Do2 must lead to tissue hypoxia.5 In reality there is a family of such Vo2/Do2 relationships with each tissue/organ having a unique Vo2/Do2 relationship and value for maximum OER that may vary with stress and disease states. Although the technology currently available makes it impracticable to determine these organ specific relationships in the critically ill patient, it is important to realise that conclusions drawn about the genesis of individual organ failure from the “global” diagram are potentially flawed.

Relationship between oxygen delivery and consumption.
In critical illness, particularly in sepsis, an altered global relationship is believed to exist (broken line DEF in fig 2⇑). The slope of maximum OER falls (DE v AB), reflecting the reduced ability of tissues to extract oxygen, and the relationship does not plateau as in the normal relationship. Hence consumption continues to increase (E–F) to “supranormal” levels of Do2, demonstrating so called “supply dependency” and the presence of a covert oxygen debt that would be relieved by further increasing Do2.6
The relationship between global Do2 and Vo2 in critically ill patients has received considerable attention over the past two decades. Shoemaker and colleagues demonstrated a relationship between Do2 and Vo2 in the early postoperative phase that had prognostic implications such that patients with higher values had an improved survival.7 A subsequent randomised placebo controlled trial in a similar group of patients showed improved survival if the values for Do2 (>600 ml/min/m2) and Svo2 (>70%) that had been achieved by the survivors in the earlier study were set as therapeutic targets (“goal directed therapy”).8
This evidence encouraged the use of “goal directed therapy” in patients with established (“late”) septic shock and organ dysfunction in the belief that this strategy would increase Vo2 and prevent multiple organ failure. Do2 was increased using vigorous intravenous fluid loading and inotropes, usually dobutamine. The mathematical linkage caused by calculating both Vo2 and Do2 using common measurements of Qt and Cao23 and the “physiological” linkage resulting from the metabolic effects of inotropes increasing both Vo2 and Do2 were confounding factors in many of these studies.9 This approach was also responsible for a considerable increase in the use of pulmonary artery catheters to direct treatment. However, after a decade of conflicting evidence from numerous small, often methodologically flawed studies, two major randomised controlled studies finally showed that there was no benefit and possibly harm from applying this approach in patients with established “shock”.10,11 Interestingly, these studies also found that those patients who neither increased their Do2 spontaneously nor in response to treatment had a particularly poor outcome. This suggested that patients with late “shock” had “poor physiological reserve” with myocardial and other organ failure caused by fundamental cellular dysfunction. These changes would be unresponsive to Shoemaker's goals that had been successful in “early” shock. Indeed, one might predict that, in patients with the increased endothelial permeability and myocardial dysfunction that typifies late “shock”, aggressive fluid loading would produce widespread tissue oedema impairing both pulmonary gas exchange and tissue oxygen diffusion. The reported increase in mortality associated with the use of pulmonary artery catheters12 may reflect the adverse effects of their use in attempting to achieve supranormal levels of Do2.
SHOULD GOAL DIRECTED THERAPY BE ABANDONED?
Recent studies examining perioperative “optimisation” in patients, many of whom also had significant pre-existing cardiopulmonary dysfunction, have confirmed that identifying and treating volume depletion and poor myocardial performance at an early stage is beneficial.13–,16 This was the message from Shoemaker's studies 20 years ago, but unfortunately it was overinterpreted and applied to inappropriate patient populations causing the confusion that has only recently been resolved. Thus, adequate volume replacement in relatively volume depleted perioperative patients is entirely appropriate. However, the strategy of using aggressive fluid replacement and vasoactive agents in pursuit of supranormal “global” goals does not improve survival in patients presenting late with incipient or established multiorgan failure.
This saga highlights the difference between “early” and “late” shock and the concept well known to traumatologists as the “golden hour”. Of the various forms of circulatory shock, two distinct groups can be defined: those with hypovolaemic, cardiogenic, and obstructive forms of shock (group 1) have the primary problem of a low cardiac output impairing Do2; those with septic, anaphylactic, and neurogenic shock (group 2) have a problem with the distribution of Do2 between and within organs—that is, abnormalities of regional Do2 in addition to any impairment of global Do2. Sepsis is also associated with cellular/metabolic defects that impair the uptake and utilisation of oxygen by cells. Prompt effective treatment of “early” shock may prevent progression to “late” shock and organ failure. In group 1 the peripheral circulatory response is physiologically appropriate and, if the global problem is corrected by intravenous fluid administration, improvement in myocardial function or relief of the obstruction, the peripheral tissue consequences of prolonged inadequacy of global Do2 will not develop. However, if there is delay in instituting effective treatment, then shock becomes established and organ failure supervenes. Once this late stage has been reached, manipulation of the “global” or convective components of Do2 alone will be ineffective. Global Do2 should nonetheless be maintained by fluid resuscitation to correct hypovolaemia and inotropes to support myocardial dysfunction.
REGIONAL OXYGEN DELIVERY
Hypoxia in specific organs is often the result of disordered regional distribution of blood flow both between and within organs rather than inadequacy of global Do2.17 The importance of regional factors in determining tissue oxygenation should not be surprising since, under physiological conditions of metabolic demand such as exercise, alterations in local vascular tone ensure the necessary increase in regional and overall blood flow—that is, “consumption drives delivery”. It is therefore important to distinguish between global and regional Do2 when considering the cause of tissue hypoxia in specific organs. Loss of normal autoregulation in response to humoral factors during sepsis or prolonged hypotension can cause severe “shunting” and tissue hypoxia despite both global Do2 and Svo2 being normal or raised.18 In these circumstances, improving peripheral distribution and cellular oxygen utilisation will be more effective than further increasing global Do2. Regional and microcirculatory distribution of cardiac output is determined by a complex interaction of endothelial, neural, metabolic, and pharmacological factors. In health, many of these processes have been intensively investigated and well reviewed elsewhere.19
Until recently the endothelium had been perceived as an inert barrier but it is now realised that it has a profound effect on vascular homeostasis, acting as a dynamic interface between the underlying tissue and the many components of flowing blood. In concert with other vessel wall cells, the endothelium not only maintains a physical barrier between the blood and body tissues but also modulates leucocyte migration, angiogenesis, coagulation, and vascular tone through the release of both constrictor (endothelin) and relaxing factors (nitric oxide, prostacyclin, adenosine).20 The differential release of such factors has an important role in controlling the distribution of regional blood flow during both health and critical illness. The endothelium is both exposed to and itself produces many inflammatory mediators that influence vascular tone and other aspects of endothelial function. For example, nitric oxide production is increased in septic shock following induction of nitric oxide synthase in the vessel wall. Inhibition of nitric oxide synthesis increased vascular resistance and systemic blood pressure in patients with septic shock, but no outcome benefit could be demonstrated.21 Similarly, capillary microthrombosis following endothelial damage and neutrophil activation is probably a more common cause of local tissue hypoxia than arterial hypoxaemia (fig 3⇓). Manipulation of the coagulation system, for example, using activated protein C may reduce this thrombotic tendency and improve outcome as shown in a recent randomised, placebo controlled, multicentre study in patients with severe sepsis.22

Example of tissue ischaemia and necrosis from extensive microvascular and macrovascular occlusion in a patient with severe meningococcal sepsis.
The clinical implications of disordered regional blood flow distribution vary considerably with the underlying pathological process. In the critically ill patient splanchnic perfusion is reduced by the release of endogenous vasoconstrictors and the gut mucosa is frequently further compromised by failure to maintain enteral nutrition. In sepsis and experimental endotoxaemia the oxygen extraction ratio is reduced and the critical Do2 increased to a greater extent in splanchnic tissue than in skeletal muscle.23 This tendency to splanchnic ischaemia renders the gut mucosa “leaky”, allowing translocation of endotoxin and possibly bacteria into the portal circulation. This toxic load may overwhelm hepatic clearance producing widespread endothelial damage. Treatment aimed at maintaining or improving splanchnic perfusion reduces the incidence of multiple organ failure and mortality.24
Although increasing global Do2 may improve blood flow to regionally hypoxic tissues by raising blood flow through all capillary beds, this is an inefficient process and, if achieved using vasoactive drugs, may adversely affect regional distribution, particularly to the kidneys and splanchnic beds. The potent α receptor agonist noradrenaline is frequently used to counteract sepsis induced vasodilation and hypotension. The increase in blood pressure may improve perfusion to certain hypoxia sensitive vital organs but may also compromise blood flow to other organs, particularly the splanchnic bed. The role of vasodilators is less well defined: tissue perfusion is frequently already compromised by systemic hypotension and a reduced systemic vascular resistance, and their effect on regional distribution is unpredictable and may impair blood flow to vital organs despite increasing global Do2. In a group of critically ill patients prostacyclin increased both Do2 and Vo2 and this was interpreted as indicating that there was a previously unidentified oxygen debt. However, there is no convincing evidence that vasodilators improve outcome in critically ill patients. An alternative strategy that attempts to redirect blood flow from overperfused non-essential tissues such as skin and muscle tissues to underperfused “vital” organs by exploiting the differences in receptor population and density between different arteries is theoretically attractive. While dobutamine may reduce splanchnic perfusion, dopexamine hydrochloride has dopaminergic and β-adrenergic but no α-adrenergic effects and may selectively increase renal and splanchnic blood flow.25
OXYGEN TRANSPORT FROM CAPILLARY BLOOD TO INDIVIDUAL CELLS
The delivery of oxygen from capillary blood to the cell depends on:
factors that influence diffusion (fig 4⇓);
the rate of oxygen delivery to the capillary (Do2);
the position of the oxygen-haemoglobin dissociation relationship (P50);
the rate of cellular oxygen utilisation and uptake (Vo2).

Diagram showing the importance of local capillary oxygen tension and diffusion distance in determining the rate of oxygen delivery and the intracellular Po2. On the left there is a low capillary Po2 and pressure gradient for oxygen diffusion with an increased diffusion distance resulting in low intracellular and mitochondrial Po2. On the right the higher Po2 pressure gradient and the shorter diffusion distance result in significantly higher intracellular Po2 values.
The sigmoid oxygen-haemoglobin dissociation relationship is influenced by various physicochemical factors and its position is defined by the Pao2 at which 50% of the Hb is saturated (P50), normally 3.5 kPa. An increase in P50 or rightward shift in this relationship reduces the Hb saturation (Sao2) for any given Pao2, thereby increasing tissue oxygen availability. This is caused by pyrexia, acidosis, and an increase in intracellular phosphate, notably 2,3-diphosphoglycerate (2,3-DPG). The importance of correcting hypophosphataemia, often found in diabetic ketoacidosis and sepsis, is frequently overlooked.26
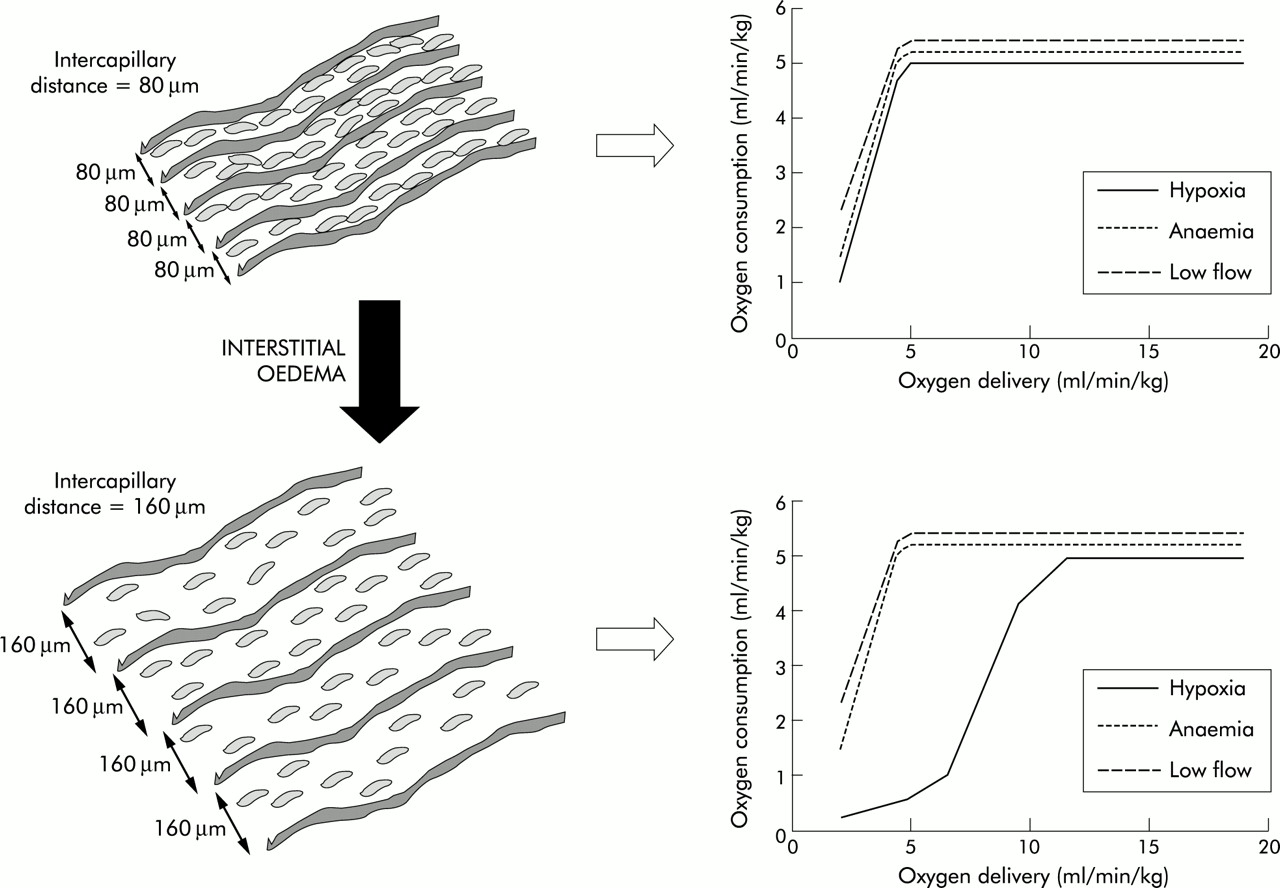
Mathematical models of tissue hypoxia show that the fall in cellular oxygen resulting from an increase in intercapillary distance is more severe if the reduction in tissue Do2 is caused by “hypoxic” hypoxia (a fall in Pao2) rather than “stagnant” (a fall in flow) or “anaemic” hypoxia (fig 5⇓).27 Studies in patients with hypoxaemic respiratory failure have also shown that it is Pao2 rather than Do2—that is, diffusion rather than convection—that has the major influence on outcome.9

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Influence of intercapillary distance on the effects of hypoxia, anaemia, and low flow on the oxygen delivery-consumption relationship. With a normal intercapillary distance illustrated in the top panels the Do2/Vo2 relationship is the same for all interventions. However, in the lower panels an increased intercapillary distance, as would occur with tissue oedema, reducing Do2 by progressive falls in arterial oxygen tension results in a change in the Do2/Vo2 relationship with Vo2 falling at much higher levels of global Do2. This altered relationship is not seen when Do2 is reduced by anaemia or low blood flow.
Thus, tissue oedema due to increased vascular permeability or excessive fluid loading may result in impaired oxygen diffusion and cellular hypoxia, particularly in clinical situations associated with arterial hypoxaemia. In these situations, avoiding tissue oedema may improve tissue oxygenation.
OXYGEN DELIVERY AT THE TISSUE LEVEL
Individual organs and cells vary considerably in their sensitivity to hypoxia.28 Neurons, cardiomyocytes, and renal tubular cells are exquisitely sensitive to a sudden reduction in oxygen supply and are unable to survive sustained periods of hypoxia, although ischaemic preconditioning does increase tolerance to hypoxia. Following complete cessation of cerebral perfusion, nuclear magnetic resonance (NMR) measurements show a 50% decrease in cellular adenosine triphosphate (ATP) within 30 seconds and irreversible damage occurs within 3 minutes. Mechanisms have developed in other tissues to survive longer without oxygen: the kidneys and liver can tolerate 15–20 minutes of total hypoxia, skeletal muscle 60–90 minutes, and vascular smooth muscle 24–72 hours. The most extreme example of hypoxic tolerance is that of hair and nails which continue to grow for several days after death.
Variation in tissue tolerance to hypoxia has important clinical implications. In an emergency, maintenance of blood flow to the most hypoxia sensitive organs should be the primary goal. Hypoxic brain damage after cardiorespiratory collapse will leave a patient incapable of independent life even if the other organ systems survive. Although tissue death may not occur as rapidly in less oxygen sensitive tissues, prolonged failure to make the diagnosis has equally serious consequences. For example, skeletal muscle may survive severe ischaemia for several hours but failure to remove the causative arterial embolus will result in muscle necrosis with the release into the circulation of myoglobin and other toxins and activation of the inflammatory response.
Tolerance to hypoxia differs in health and disease. In a septic patient inhibition of enzyme systems and oxygen utilisation reduces hypoxic tolerance.29 Methods aimed at enhancing metabolic performance including the use of alternative substrates, techniques to inhibit endotoxin induced cellular damage, and drugs to reduce oxidant induced intracellular damage are currently under investigation. Ischaemic preconditioning of the heart and skeletal muscle is recognised both in vivo and in experimental models. Progressive or repeated exposure to hypoxia enhances tissue tolerance to oxygen deprivation in much the same way as altitude acclimatisation. An acclimatised mountaineer at the peak of Mount Everest can tolerate a Pao2 of 4–4.5 kPa for several hours, which would result in loss of consciousness within a few minutes in a normal subject at sea level.
What is the critical level of tissue oxygenation below which cellular damage will occur? The answer mainly depends on the patient's circumstances, comorbid factors, and the duration of hypoxia. For example, young previously healthy patients with the acute respiratory distress syndrome tolerate prolonged hypoxaemia with saturations as low as 85% and can recover completely. In the older patient with widespread atheroma, however, prolonged hypoxaemia at such levels would be unacceptable.
RECOGNITION OF INADEQUATE TISSUE OXYGEN DELIVERY
The blood lactate concentration is an unreliable indicator of tissue hypoxia. It represents a balance between tissue production and consumption by hepatic and, to a lesser extent, by cardiac and skeletal muscle.30 It may be raised or normal during hypoxia because the metabolic pathways utilising glucose during aerobic metabolism may be blocked at several points.31 Inhibition of phosphofructokinase blocks glucose utilisation without an increase in lactate concentration. In contrast, endotoxin and sepsis may inactivate pyruvate dehydrogenase, preventing pyruvate utilisation in the Krebs cycle resulting in lactate production in the absence of hypoxia.32 Similarly, a normal Do2 with an unfavourable cellular redox state may result in a high lactate concentration, whereas compensatory reductions in energy state [ATP]/[ADP][Pi] or [NAD+]/[NADH] may be associated with a low lactate concentration during hypoxia.33 Thus, the value of a single lactate measurement in the assessment of tissue hypoxia is limited.34 The suggestion that pathological supply dependency occurs only when blood lactate concentrations are raised is incorrect as the same relationship may be found in patients with normal lactate concentrations.35 Serial lactate measurements, particularly if corrected for pyruvate, may be of greater value.
Measurement of individual organ and tissue oxygenation is an important goal for the future. These measurements are difficult, require specialised techniques, and are not widely available. At present only near infrared spectroscopy and gastric tonometry have clinical applications in the detection of organ hypoxia.24 In the future NMR spectroscopy may allow direct non-invasive measurement of tissue energy status and oxygen utilisation.36
CELLULAR OXYGEN UTILISATION
In general, eukaryotic cells are dependent on aerobic metabolism as mitochondrial respiration offers greater efficiency for extraction of energy from glucose than anaerobic glycolysis. The maintenance of oxidative metabolism is dependent on complex but poorly understood mechanisms for microvascular oxygen distribution and cellular oxygen uptake. Teleologically, the response to reduced blood flow in a tissue is likely to have evolved as an energy conserving mechanism when substrates, particularly molecular oxygen, are scarce. Pathways that use ATP are suppressed and alternative anaerobic pathways for ATP synthesis are induced.37 This process involves oxygen sensing and transduction mechanisms, gene activation, and protein synthesis.
CELLULAR METABOLIC RESPONSE TO HYPOXIA
Although cellular metabolic responses to hypoxia remain poorly understood, the importance of understanding and modifying the cellular responses to acute hypoxia in the critically ill patient has recently been appreciated. In isolated mitochondria the partial pressure of oxygen required to generate high energy phosphate bonds (ATP) that maintain aerobic cellular biochemical functions is only about 0.2–0.4 kPa.17,28 However, in intact cell preparations hypoxia induced damage may result from failure of energy dependent membrane ion channels with subsequent loss of membrane integrity, changes in cellular calcium homeostasis, and oxygen dependent changes in cellular enzyme activity.28 The sensitivity of an enzyme to hypoxia is a function of its Po2 in mm Hg at which the enzyme rate is half maximum (Kmo2),28 and the wide range of values for a variety of cellular enzymes is shown in table 2⇓, illustrating that certain metabolic functions are much more sensitive to hypoxia than others. Cellular tolerance to hypoxia may involve “hibernation” strategies that reduce metabolic rate, increased oxygen extraction from surrounding tissues, and enzyme adaptations that allow continuing metabolism at low partial pressures of oxygen.37
Oxygen affinities of cellular enzymes expressed as the partial pressure of oxygen in mm Hg at which the enzyme rate is half maximum (Kmo2)
Anaerobic metabolism is important for survival in some tissues despite its inherent inefficiency: skeletal muscle increases glucose uptake by 600% during hypoxia and bladder smooth muscle can generate up to 60% of total energy requirement by anaerobic glycolysis.38 In cardiac cells anaerobic glucose utilisation protects cell membrane integrity by maintaining energy dependent K+ channels.39 During hypoxic stress endothelial and vascular smooth muscle cells increase glucose transport through the expression of membrane glucose transporters (GLUT-1 and GLUT-4) and the production of glycolytic enzymes, thereby increasing anaerobic glycolysis and maintaining energy production.38 High energy functions like ion transport and protein production are downregulated to balance supply and demand.
Cellular oxygen utilisation is inhibited by metabolic poisons (cyanide) and toxins associated with sepsis such as endotoxin and other cytokines, thereby reducing energy production.29 It is yet to be established whether there are important differences in the response to tissue hypoxia resulting from damage to mitochondrial and other intracellular functions as occurs in poisoning and sepsis, as opposed to situations such as exercise and altitude when oxygen consumption exceeds supply.
OXYGEN SENSING AND GENE ACTIVATION
The molecular basis for oxygen sensing has not been established and may differ between tissues. Current evidence suggests that, following activation of a “hypoxic sensor”, the signal is transmitted through the cell by second messengers which then activate regulatory protein complexes termed transcription factors.40,41 These factors translocate to the nucleus and bind with specific DNA sequences, activating various genes with the subsequent production of effector proteins. It has long been postulated that the “hypoxic sensor” may involve haem-containing proteins, redox potential or mitochondrial cytochromes.42 Recent evidence from vascular smooth muscle suggests that hypoxia induced inhibition of electron transfer at complex III in the electron transport chain may act as the “hypoxic sensor”.43 This sensing mechanism is associated with the production of oxygen free radicals (ubiquinone cycle) that may act as second messengers in the activation of transcription factors.
Several transcription factors play a role in the response to tissue hypoxia including hypoxia inducible factor 1 (HIF-1), early growth response 1 (Erg-1), activator protein 1 (AP-1), nuclear factor kappa-B (NF-χB), and nuclear factor IL-6 (NF-IL-6). HIF-1 influences vascular homeostasis during hypoxia by activating the genes for erythropoietin, nitric oxide synthase, vascular endothelial growth factor, and glycolytic enzymes and glucose transport thereby altering metabolic function.40 Erg-1 protein is also rapidly induced by hypoxia leading to transcription of tissue factor, which triggers prothrombotic events.41
Key points
Restoration of global oxygen delivery is an important goal in early resuscitation but thereafter circulatory manipulation to sustain “supranormal” oxygen delivery does not improve survival and may be harmful.
Regional distribution of oxygen delivery is vital: if skin and muscle receive high blood flows but the splanchnic bed does not, the gut may become hypoxic despite high global oxygen delivery.
Microcirculatory, tissue diffusion, and cellular factors influence the oxygen status of the cell and global measurements may fail to identify local tissue hypoxaemia.
Supranormal levels of oxygen delivery cannot compensate for diffusion problems between capillary and cell, nor for metabolic failure within the cell.
When assessing Do2/Vo2 relationships, direct measurements should be used to avoid errors due to mathematical linkage.
Strategies to reduce metabolic rate to improve tissue oxygenation should be considered.