Article Text

Abstract
Background: Hypochlorous acid (HOCl) is the main oxidant of activated polymorphonuclear neutrophil granulocytes (PMN) and generated by myeloperoxidase during respiratory burst. This study investigates the effects of HOCl on pulmonary artery pressure (PAP) and vascular permeability and characterises the influence of arachidonic acid (AA) and eicosapentaenoic acid (EPA) on the observed effects.
Methods: HOCl (500, 1000, 2000 nmol/min) was continuously infused into the perfusate (Krebs-Henseleit buffer solution, KHB). AA or EPA in subthreshold doses (both 2 nmol/min) or buffer were simultaneously infused using a separate port. PAP, pulmonary venous pressure (PVP), ventilation pressure, and lung weight gain were continuously recorded. The capillary filtration coefficient (Kf,c) was calculated before and 30, 60, and 90 minutes after starting the HOCl infusion.
Results: HOCl application resulted in a dose dependent increase in PAP and Kf,c. The onset of these changes was inversely related to the HOCl dose used. The combined infusion of AA with HOCl resulted in a significant additional rise in pressure and oedema formation which forced premature termination of all experiments. The combination of EPA with HOCl did not result in an enhancement of the HOCl induced rise in pressure and oedema formation.
Conclusions: Changes in the pulmonary microvasculature caused by HOCl are differently influenced by ω-6 and ω-3 polyunsaturated free fatty acids, suggesting a link between neutrophil derived oxidative stress and pulmonary eicosanoid metabolism.
- acute lung injury
- oxidative stress
- arachidonic acid
- eisosapentaenoic acid
Statistics from Altmetric.com
Although states of acute lung vascular injury such as the adult (ARDS) and infant respiratory distress syndrome (IRDS) are also observed under neutropenic conditions—for example, in patients with chemotherapy induced aplasia—neutrophil granulocytes (PMN) are regarded as key cells for aggravation and perpetuation of pulmonary inflammatory events. Activated neutrophils affect surrounding lung tissue via several potentially pathogenic cellular mechanisms including the release of lysosomal proteolytic enzymes, the production of prostanoids, and the generation of highly reactive oxygen radicals and intermediates.
Hypochlorous acid (HOCl) is the main oxidant generated by activated neutrophils. It is synthesised by a myeloperoxidase (MPO) mediated reaction from hydrogen peroxide (H2O2) and chloride and is much more reactive than the latter oxidant.1 Because its synthesis depends on neutrophil released MPO, HOCl is a neutrophil specific oxidant. HOCl as a non-radical oxidant causes oxidative modification of free functional groups2 and subsequently functional alterations of proteins.3 Moreover, we found comparable vascular effects of both stimulated neutrophil granulocytes and HOCl on the circulation of isolated perfused rabbit lung preparations.4,5 We therefore consider that HOCl, which is not expected to cause non-specific effects or cell damage due to lipid peroxidation if used in non-cytolytic doses, is a useful model for the investigation of the effects of neutrophil derived oxidative stress in isolated organ models.
Only a few data are available on the biological effects of non-radical oxidants such as H2O2 and HOCl on the level of cells or organs, especially the lungs. In most experiments HOCl is characterised by its cytolytical action to different target cells in vitro.6 Some more recent publications have studied the effects of non-cytolytic doses of superoxide anion,7 H2O2,8 and HOCl,9 and there is some evidence that oxidative stress may activate pulmonary arachidonic acid metabolism predominantly via cyclooxygenase pathways. Nevertheless, there are no data which characterise qualitatively and quantitatively the effect of non-cytolytic doses of HOCl on the pulmonary microvasculature in an isolated organ model.
Parenteral nutrition in critically ill patients with ARDS is common, and free fatty acid pools are augmented in states of parenteral nutrition using lipid emulsions in these patients under clinical conditions.10 Furthermore, a rise in pulmonary artery pressure (PAP) and hydrostatic oedema formation due to consecutively augmented right ventricular afterload was observed after infusion of soybean or safflower based lipid emulsions which have been attributed to metabolites generated from liberated precursors, particularly arachidonic acid (AA).11 Synthetic lipid aggregates are known to activate endothelial lipoprotein lipase followed by a translocation of this enzyme into the vascular compartment with a resultant increase in plasma free fatty acids. This effect is additionally augmented by the infusion of heparin, used in most critically ill patients.12
Against this background, we designed a study to investigate the effect of continuous HOCl infusion in different non-cytolytic dosages on the PAP response and vascular permeability. We also studied the impact of pulmonary AA metabolism on the HOCl induced pulmonary changes by continuous additional administration of subthreshold doses of polyunsaturated free fatty acids (PUFAs)—that is, the proinflammatory AA and its natural antagonist eicosapentaenoic acid (EPA)—both of which are well known substrates of pulmonary AA metabolism.
METHODS
Reagents
The C20:4 ω-6 free fatty acid AA and the C20:5 ω-3 free fatty acid EPA were purchased from Sigma (München, Germany). Sterile, pyrogen free Krebs-Henseleit buffer solution (KHB) was obtained from Serag-Wiesner (Naila, Germany) and was used without colloid. All other biochemical reagents were obtained from Sigma and were used in para analysis quality. The concentration of the HOCl stock solution was determined spectrophotometrically (ε290 nm = 350/mol/cm) immediately before use. HOCl was obtained from Sigma.
Isolated rabbit lung model: general procedure
The preparation of isolated rabbit lungs was originally described by Starling and Knowlton in 1912 and used with some modifications. Briefly, rabbits of either sex of body weight 2.5–3.5 kg were used. The animals were deeply anaesthetised with a mixture of 30 mg ketamine base and 27 mg xylazine and heparinised with 1000 U heparin per kg. The rabbits were ventilated after tracheotomy with 4% CO2, 17% O2, and 79% N2 (tidal volume 9 ml/kg; frequency 30/min). Thoracotomy was performed and a wide bore cannula was inserted under flow into the pulmonary artery via the right ventricular outflow tract. Ice cold KHB was used for perfusion with an initial flow of 10–20 ml/min in order to avoid possible biochemical events stimulated by the change of perfusion fluid. After removal from the thorax the lungs were placed in a 4°C equilibrated chamber, freely suspended from a force transducer. A second cannula was placed in the left ventricle after removal of the leaflets of the mitral valve. The left atrium was ligated. In combination, the temperature of the perfusion fluid and of the housing chamber as well as the perfusion flow were gradually increased over 20 minutes. The flow rate was set at 100 ml/min with recirculation of the buffer medium (total circulating volume 300 ml) after extensive rinsing of the vascular bed. Formaldehyde sterilised perfusion circuit tubing and endotoxin free buffer fluids were used throughout. The endotoxin content of the recirculating perfusate was repeatedly measured in previous experiments and never reached 10 pg/ml (the detection limit of the test used).13 The buffer contained 132.8 mM NaCl, 5.2 mM KCl, 1.1 mM KH2PO4, 24.1 mM NaHCO3, 2.4 mM CaCl, 1.3 mM MgPO4, and 240 mg/dl glucose. The perfusate temperature was set at 37°C and the pH of the perfusion fluid was held constant between 7.35 and 7.45. PAP, pulmonary venous pressure (PVP, measured in the left ventricle after cardioplegia had occurred at the end of the preparation procedure), inflation pressure, and the weight gain of the isolated organ were continuously registered. After a steady state period of 45 minutes, including a hydrostatic challenge at t = –15 minutes, only lungs with no signs of leakage at the catheter insertion sites, with a homogenous white appearance and no signs of oedema formation, having lost weight during the phase of temperature increase, and being completely isogravimetric during the steady state period were selected for the study. After steady state had been reached, PAP ranged between 7 and 11 torr, inspiratory peak inflation pressure between 7 and 9 torr, and ventilation was performed without PEEP. The PVP was adjusted to 2 torr.
Gravimetric estimation of capillary filtration coefficient (Kf,c)
Kf,c (10−4 ml/s/cm H2O/g) was determined gravimetrically from the slope of lung weight gain after a sudden venous pressure increase of 7.5 torr for 8 minutes (hydrostatic challenge). Perfusion and ventilation were not interrupted during this time. The slow phase of weight gain was taken to begin 2 minutes after the increase in venous pressure. Time zero extrapolation of the slope of weight gain curve was performed using a semilogarithmic plot of the rate of lung weight gain (ΔW/Δt). Kf,c was calculated from the change in ΔW/Δt induced by the disturbance of fluid balance according to hydrostatic challenge and was related to wet weight lung (WWL) which was calculated from body weight (BW) using WWL = BW × 0.0024.
Estimation of vascular compliance
Total vascular compliance (C, g/cm H2O) is defined as the change in vascular volume per change in microvascular pressure. The total rapid change in weight over the first 1–2 minutes after onset of the venous pressure challenge was taken as pure vascular filling and used for the calculation of compliance. The tangent to the slope of weight gain 2 minutes after the onset of the increase in venous pressure was therefore elongated to time zero of the pressure increment to correct for the fluid filtration during the first 2 minutes.
Estimation of total fluid retention
The total fluid retention (ΔW, g) was determined as the remaining difference of weight before and after a hydrostatic challenge corresponding to the remaining interstitial fluid filtered during hydrostatic challenge after normalisation of venous pressure and equilibration of a new fluid balance.
Analytical methods
The concentration of potassium and lactate dehydrogenase (LDH) activity were measured by routine methods at the Institute of Clinical Chemistry of the Medizinische Universitätsklinik Würzburg using a Hitachi 747 multianalyser with commercial test kits (Boehringer Mannheim, Mannheim, Germany).
Experimental protocol
Infusion of HOCl into the arterial line of the system was started at t = 0 min after a 45 minute steady state period. HOCl, which was diluted to different concentrations with perfusate, was infused with a constant flow rate of 0.5 ml/min so that HOCl doses of 500 (HOCl 500 group), 1000 (HOCl 1000 group), and 2000 nmol/min (HOCl 2000 group) were administered. Hydrostatic challenges were performed at 30, 60, and 90 minutes. The results were compared with a control group using buffer application alone. In two other settings infusion of PUFAs (AA and EPA) were used. Subthreshold doses of both substances were continuously infused into the perfusate starting after steady state at t = 0 min using a separate infusion port. AA was used in a dose of 2 nmol/min alone (AA group) or in combination with a continuous infusion of 500 (HOCl 500 + AA group), 1000 (HOCl 1000 + AA group), or 2000 nmol/min HOCl (HOCl 2000 + AA group). EPA was infused in a dose of 2 nmol/min alone (EPA group) or in combination with 500 (HOCl 500 + EPA group), 1000 (HOCl 1000 + EPA group), or 2000 nmol/min HOCl (HOCl 2000 + EPA group), respectively. Control studies with application of the carrier fluids alone were also performed.
Potassium concentration and LDH activity were measured immediately before hydrostatic challenges in all protocols.
Statistical methods
Data are given as mean (SE). To evaluate statistical significance, one way analysis of variance (ANOVA) was performed according to previously published work.13 Significance was assumed at p<0.05.
RESULTS
Pulmonary artery pressure response
There were no significant differences in baseline PAP (at t = 0 min) between all groups. No pressure response was observed in control experiments without HOCl application.
HOCl caused a dose dependent pressure response; the higher the dose of HOCl, the earlier the maximum pressure response occurred (fig 1) and the earlier the pressure increase started (figs 2, 3, 4). The maximum PAP in all HOCl groups differed significantly from control experiments without HOCl but the differences between the HOCl groups were small and did not reach levels of significance (fig 5).

Recording time after which experiments were terminated (tges) and after which maximum pulmonary artery pressure was reached (tPAPmax). Data are given as mean (SE). *p<0.05 versus corresponding HOCl group. HOCl=hypochlorous acid; AA=arachidonic acid; EPA=eicosapentaenoic acid.
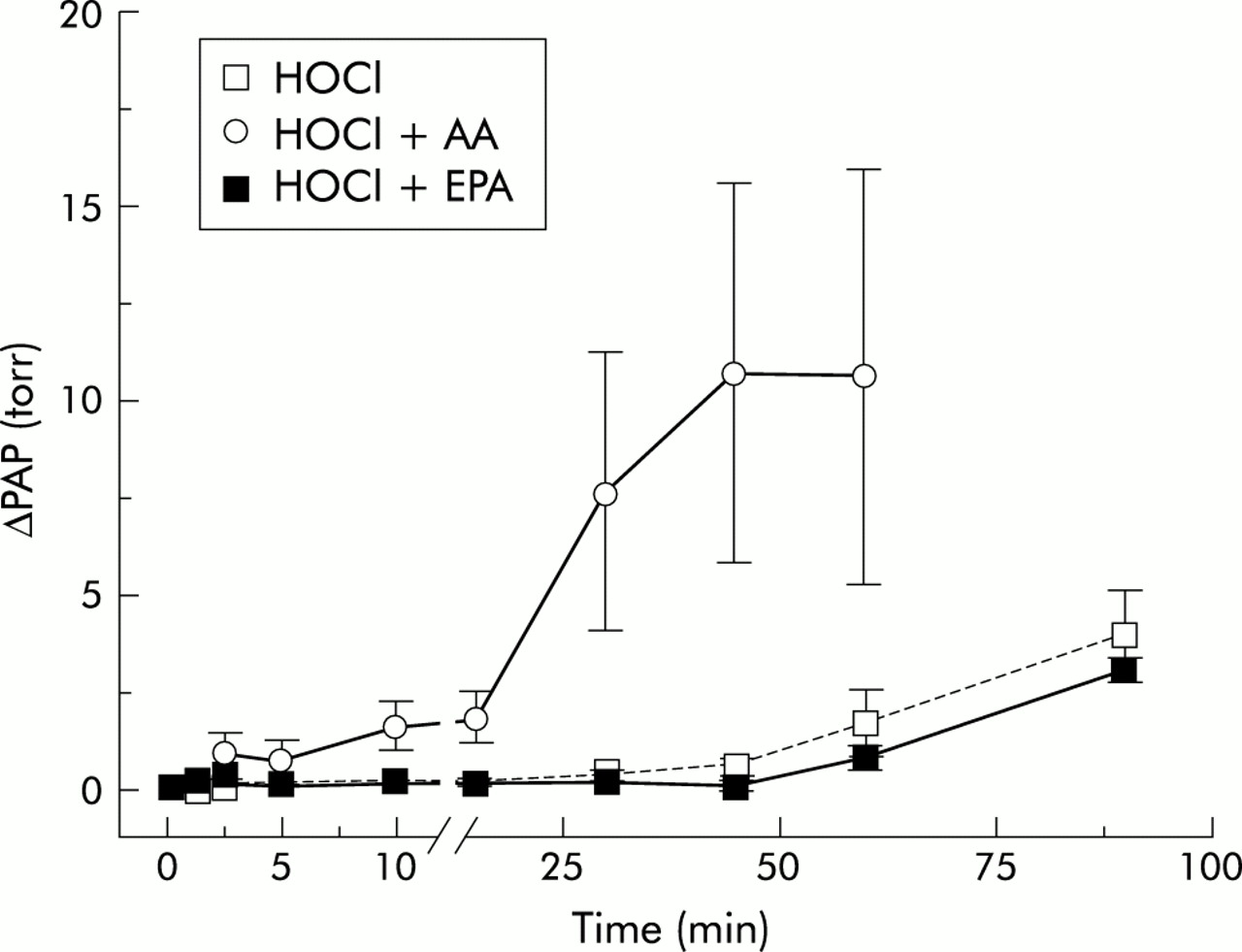
Time course of change in pulmonary artery pressure (ΔPAP) in HOCl 500 group experiments. The time course of the mean ΔPAP is plotted against the recording time. Data are given as mean (SE). HOCl=hypochlorous acid; AA=arachidonic acid; EPA=eicosapentaenoic acid.
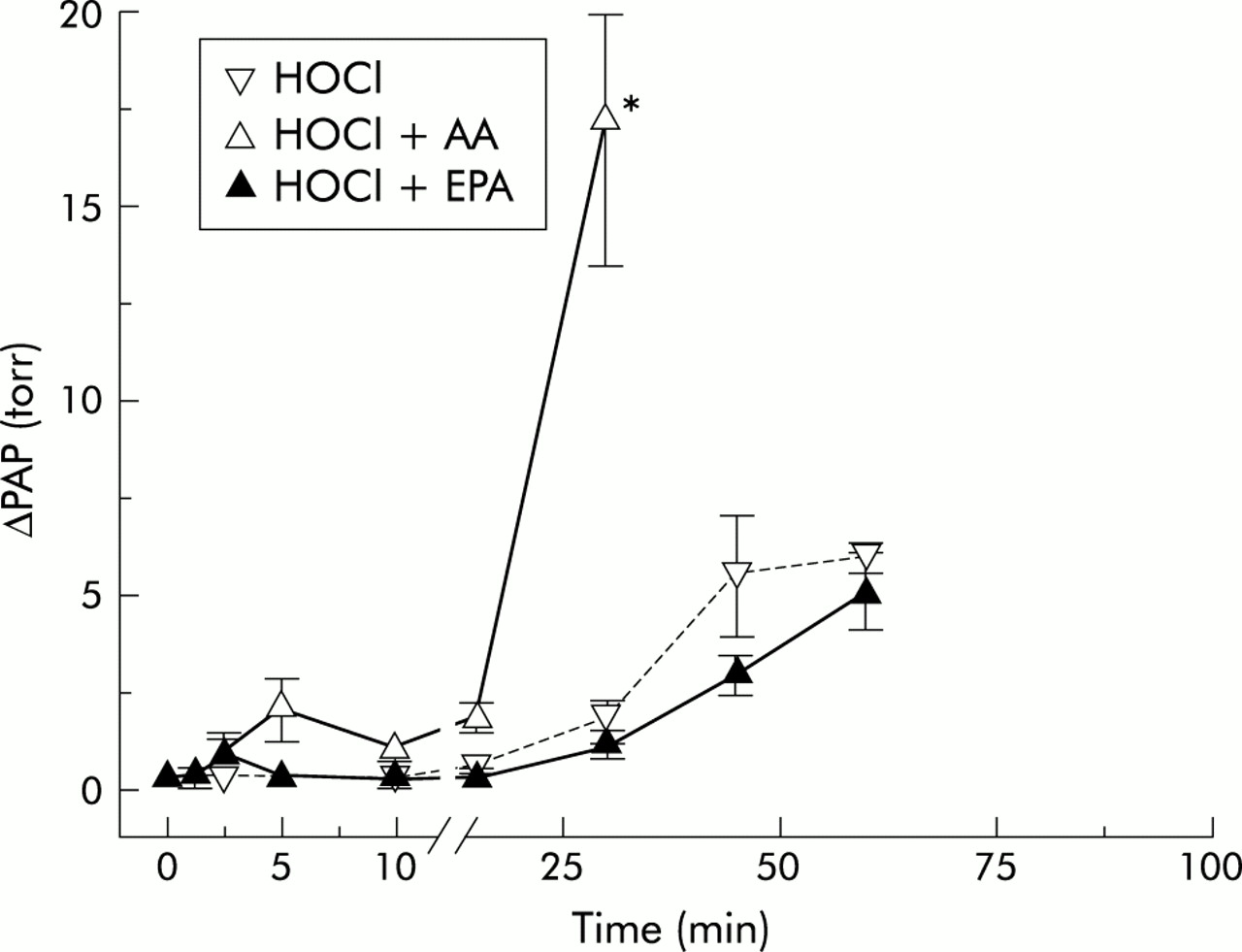
Time course of change in pulmonary artery pressure (ΔPAP) in HOCl 1000 group experiments. The time course of the mean ΔPAP is plotted against the recording time. Data are given as mean (SE). *p<0.05 versus corresponding HOCl group. HOCl=hypochlorous acid; AA=arachidonic acid; EPA=eicosapentaenoic acid.

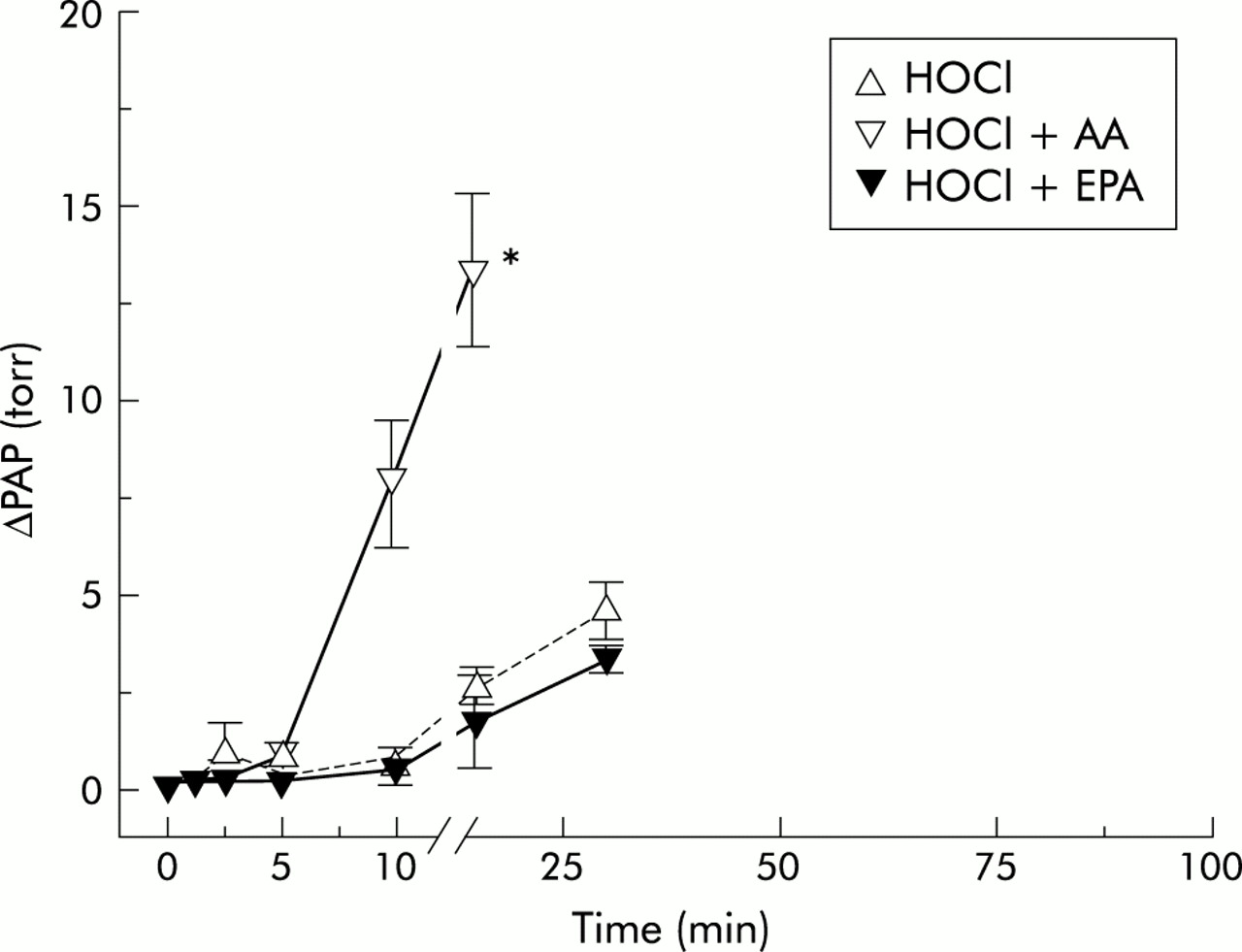
Time course of change in pulmonary artery pressure (ΔPAP) in HOCl 2000 group experiments. The time course of the mean ΔPAP is plotted against the recording time. Data are given as mean (SE). *p<0.05 versus corresponding HOCl group. HOCl=hypochlorous acid; AA=arachidonic acid; EPA=eicosapentaenoic acid.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Baseline pulmonary artery pressure after steady state (PAP(t = 0)) and maximum pulmonary artery pressure (PAPmax.). Data are given as mean (SE). *p<0.05 versus corresponding HOCl group. HOCl=hypochlorous acid; AA=arachidonic acid; EPA=eicosapentaenoic acid.
Continuous infusion of AA without HOCl had no effect on PAP at the dose used in control experiments (data not given in detail). However, when combined with HOCl it resulted in a significant additional rise in pressure which started earlier and reached a maximum earlier than in the groups given HOCl alone (fig 1), and amounted to a rise in maximum pressure nearly three times that achieved with application of HOCl alone (fig 5). ΔPAP began to rise at 10 minutes and reached its maximum approximately 60 minutes later in the HOCl 500 + AA group (figs 1 and 2). In the HOCl 1000 + AA group the rise in PAP started at 5 minutes and ΔPAP was significantly higher than in the corresponding HOCl group at 30 minutes (fig 3). In the HOCl 2000 + AA group the increase in ΔPAP was significant compared with the corresponding HOCl group at 10 minutes and the maximum pressure response was nearly reached at 15 minutes (fig 4). In all three groups PAPmax was significantly higher than in the corresponding HOCl groups.
Continuous infusion of EPA also had no effect on PAP in control experiments without HOCl (data not given in detail). However, unlike AA, EPA did not have any additional effect on the observed pressure response when applied with HOCl. ΔPAP was reduced by additional EPA in all three EPA-HOCl groups compared with the corresponding groups given HOCl alone, even if levels of significance were not reached (figs 2–4). PAPmax was also reduced in all three HOCl-EPA groups, but the differences were not significant.
Vascular permeability
Table 1 shows the results of hydrostatic challenge. There were no significant differences in the baseline values of vascular compliance (Kf,c) and fluid retention between the groups.
Data calculated from hydrostatic challenges
In control experiments without HOCl there were no changes in vascular permeability and compliance during the whole study. Continuous application of 500 nmol/min HOCl caused no significant increase in Kf,c and fluid retention compared with control experiments. However, an increase in vascular permeability was observed in the HOCl 1000 group with about a sixfold increase in Kf,c and a 17-fold increase in fluid retention, both reaching levels of significance after 60 minutes. Severe oedema finally resulted in premature termination of the experiments after 73 (3.4) minutes (fig 1). Infusion of 2000 nmol/min HOCl caused severe oedema with an increase in Kf,c of about ninefold and an increase in fluid retention of nearly 30-fold, both reaching levels of significance after 30 minutes. The experiments had to be terminated after 44 (3.9) minutes (fig 1).
In control experiments without HOCl, infusion of AA caused no significant changes in vascular permeability (indicated by Kf,c and fluid retention) or in vascular compliance. However, simultaneous infusion of AA and HOCl led to a significant enhancement of the HOCl induced effects on vascular permeability. In the HOCl 500 + AA group severe oedema developed shortly after the first hydrostatic challenge at 30 minutes and the experiments had to be terminated after 83 (8.5) minutes because of severe interstitial and intra-alveolar oedema (fig 1). In the HOCl 1000 + AA group oedema formation had already started to develop at 30 minutes, as indicated by a nearly fourfold increase in Kf,c and an increase in fluid retention of about 11-fold in the first hydrostatic challenge. Immediately after the first hydrostatic challenge oedema reached a maximum and the experiments had to be terminated at 43 (6.5) minutes (fig 1). In the HOCl 2000 + AA group oedema started to develop immediately after infusion of both stimuli had started and the experiments had to be terminated after only 25 (2.2) minutes because of severe oedema; this meant a significant difference in the mean recording time compared with the corresponding HOCl group without additional AA (fig 1).
Infusion of EPA caused no significant chances in vascular permeability and vascular compliance in control experiments without HOCl (table 1). In contrast to the findings with AA, combined application of EPA and HOCl did not result in an enhancement of HOCl induced oedema formation. In the HOCl 500 + EPA group the values for Kf,c and fluid retention were almost identical during the whole recording time. In the HOCl 1000 + EPA group oedema formation, characterised by a rise in Kf,c of about fourfold and an 11-fold rise in fluid retention, was nearly the same as in the corresponding HOCl group without EPA. The same result was seen in the HOCl 2000 + EPA group in which severe oedema formation occurred during the first hydrostatic challenge, as was seen in the corresponding HOCl group without EPA. Furthermore, in all experimental groups with EPA application the mean recording time was not significantly different from the corresponding HOCl groups (fig 1).
Vascular compliance
The values for vascular compliance were constant or even decreased during the whole period of study in all experiments (table 1).
Potassium and LDH release
LDH and potassium measured from repeated perfusate probes were not significantly changed during the whole period of the study in all experiments.
DISCUSSION
This study characterises the qualitative and quantitative effects of continuously infused AA and EPA on HOCl induced acute vascular injury in isolated buffer perfused rabbit lungs. HOCl causes a significant rise in vascular resistance, as indicated by a dose dependent increase in PAP, and a significant increase in vascular permeability as indicated by a dose dependent rise in capillary filtration coefficient (Kf,c) and fluid retention (ΔW). These effects were significantly augmented by the addition of AA, whereas the addition of EPA caused no adverse effects in this model of oxidative stress induced lung injury.
HOCl induced lung injury
In addition to their main oxidant HOCl, PMN release H2O2 and superoxide anion. Furthermore, the interaction of the two latter oxidants leads to formation of hydroxyl radicals in the presence of (mostly iron based) metal catalysts. However, the biological importance of these metabolites may be limited in their ability to cause functional alterations for a number of reasons.
Superoxide anion radical spontaneously dismutates to H2O2 in a very rapid reaction and is therefore not available in suitable quantities for reactions with other targets. H2O2 is a stable oxidant. However, most of this oxidant is consumed by neutrophils themselves to generate HOCl in an MPO mediated reaction. For this reason, only small quantities of H2O2 are detectable in the extracellular pool.14 In addition, the catalytic formation of hydroxyl radical is prevented by iron binding lactoferrin released by stimulated PMN.15
In contrast, HOCl is an extremely powerful oxidant generated in high quantities. It is also reported to attack a wide range of targets and, under non-specific reactions without biological responses, functional changes at the level of cellular mediator generation should be expected.
The dosages used ranged from 500 nmol/min to 2000 nmol/min, corresponding to perfusate concentrations of 5–20 μM in the arterial line which are regarded as very low for the following reasons. As is known from in vitro experiments, 106 PMN release 2 × 10−7 mol HOCl in 2 hours following maximum stimulation,16 an HOCl release of 1.67 nmol/min. 3 × 108 maximum stimulated neutrophils were needed to achieve the release of 500 nmol/min HOCl. If one theoretically assumes that the cells would be homogeneously distributed to a recirculating perfusate volume of 300 ml as used in our experiments, a cell count of 1000/μl activated neutrophils corresponds with this synthesis rate. Activated PMN in perfusate cell counts of 2000–4000/μl were therefore needed to get releasing rates of 1000 and 2000 nmol/min, as used in our experiments.
Corresponding to previously published data which have provided evidence for a close relationship between the action of activated neutrophils and of infused HOCl in isolated rabbit lungs,4 we observed time and dose dependent changes in PAP and vascular permeability. The rise in vascular permeability became particularly dominant, whereas the PAP response was moderate. The time dependence of the vascular response, which was reciprocal to the dose of HOCl, may be explained by a depletion of cellular antioxidant mechanisms. The nearly constant product of HOCl infusion rate and the mean recording time after which oedema formation caused premature termination of the experiments emphasises this hypothesis. The changes in PAP and vascular permeability occurred without any evidence for simultaneous cell damage, as proved by the lack of LDH release into the recirculating perfusate. Even if LDH might be oxidatively inactivated by infused HOCl, there was no rise in the potassium concentration in the perfusate during the entire recording time in all experiments using HOCl, also indicating that no cell lysis occurred.
Pulmonary AA metabolism
Up to micromolar concentrations of free AA are locally synthesised during acute inflammation.17 Furthermore, studies with isolated lungs and co-cultures of different pulmonary cells have shown that intercellular exchanges of free AA exist which may contribute to transcellular eicosanoid synthesis.18 We recently published data which provide evidence for a possible link between the action of neutrophil derived HOCl and pulmonary eicosanoid metabolism using inhibitors of cyclooxygenase and 5-lipoxygenase.5,19 The generation of cyclooxygenase metabolites as possible mediators of non-cytolytic oxidative effects after respiratory burst has also been described for models of oxidative stress other than HOCl (xanthine oxidase system, H2O2). Respiratory epithelial cells generate prostaglandin (PG) F2α,20 the responsiveness of isolated canine bronchi is modified by the generation of PGE2 and I2,21 and in pig lungs vasoconstriction occurs according to the synthesis of thromboxane and prostacyclin.7 The effect of oxidative stress is attenuated by different cyclooxygenase inhibitors indicating an important role of AA and its cyclooxygenase metabolites as mediators of injury in all these experiments.
The results of our experiments are consistent with these findings. The described adverse effects of HOCl with consecutive severe vascular injury are significantly aggravated by additional AA, whereas the rise in PAP and vascular permeability remains nearly unchanged or is even slightly diminished when EPA is administered.
In patients with psoriasis, serum levels of about 4 μmol/l AA and 0.5–1 μmol/1 EPA were found. After infusion of conventional ω-6 based lipid emulsions, AA concentrations up to 8 μmol/l were measured. With ω-3 enriched preparations EPA concentrations of 2 and 4 μmol/l were achieved.22 In our experiments with continuous PUFA infusions cumulative doses up to 210 nmol were added during the 105 minute recording time which correspond to maximum perfusate concentrations up to 0.7 μmol/l. Perfusate levels achieved with both AA and EPA do not therefore exceed serum levels measured after infusion of lipid emulsions under clinical conditions.
The observed effects can be explained by the synthesis of less active EPA derived eicosanoids. A direct anti-oxidative effect of EPA, as described for lipids in general, cannot be completely ruled out in our experiments. However, since the structural differences between the two free fatty acids are minimal, it is very unlikely that the difference in activity of EPA and AA is caused by EPA acting as a scavenger. Furthermore, the addition of AA also produced adverse effects in non-oxidative stress induced lung injury models.13,23 Against this background, our results suggest that even subthreshold concentrations of AA may produce adverse effects in states of acute pulmonary inflammation.
EPA is not usually detected in membrane phospholipids of most cells because western diets contain almost no EPA rich ω-3 lipids. It has been shown that it is possible to influence the relation between the EPA and AA content of different blood cells in healthy individuals toward the ω-3 fatty acids by the use of diets rich in fish oil.24 Fish oil derived (EPA enriched) lipid infusions possibly combine parenteral nutrition with the aim of suppressing inflammatory events. A manifold increase in plasma free EPA levels can be achieved using this strategy.25 Both the synthesis of less potent EPA derived eicosanoids and anti-inflammatory effects have been seen in several clinical and experimental settings.26,27 The beneficial effects of enteral feeding with an EPA enriched formula on pulmonary neutrophil recruitment, gas exchange, requirement for ventilation, length of stay in the intensive care unit, and the reduction of new organ failures in patients with ARDS have also been shown.28 No clinical studies have been performed on the effect of parenteral therapy in patients with acute lung injury, but experimental data are available.29,30 Experiments with a single dose application of EPA in a septic lung model using Escherichia coli haemolysin have shown that micromolar concentrations of EPA were required to demonstrate the same adverse effects achieved by 5–15 nM AA.13
The results of our experiments show distinct modifying effects of ω-6 and ω-3 fatty acids on oxidative stress induced disturbances of pulmonary vascular resistances and vascular permeability, suggesting an involvement of eicosanoid metabolism in oxidative stress induced lung injury. Both augmented oxidative stress and activation of eicosanoid metabolism are common in states of acute inflammation. Our data may in part explain some of the beneficial clinical findings of fish oil derived EPA enriched emulsions for enteral or parenteral nutrition, and may offer new therapeutic approaches to acute lung injury.
REFERENCES
Footnotes
-
This manuscript includes portions of the doctoral thesis of N Rüenauver.