Article Text

Abstract
Although restrictive lung disease is the predominant abnormality of pulmonary function in patients with thalassaemia major (TM), its aetiology and its association with pulmonary hypertension (PH) detected in some patients with TM remains unknown. We report a patient with TM, iron overload, frequent pulmonary infections, and progressive severe precapillary PH over the previous 5 years. A severe restrictive pattern and interstitial lung fibrosis were revealed by pulmonary function tests and high resolution computed tomography, respectively. This presentation suggests that interstitial fibrosis may complicate lung involvement in TM and can significantly contribute to the development of PH.
- thalassaemia major
- pulmonary hypertension
- interstitial lung fibrosis
Statistics from Altmetric.com
Impairment of cardiac function is a well documented complication of thalassaemia major (TM) attributed to haemochromatosis, and congestive heart failure resulting from decreased left ventricular systolic function is the main cause of early mortality.1 ,2 Pulmonary hypertension (PH) is relatively rare,3 ,4 especially in the absence of left ventricular dysfunction. Its aetiology remains uncertain.
The predominant abnormality of pulmonary function in TM is restrictive disease.5 ,6 Although transfusional iron burden and increasing age seem to play a role,5 the nature of this restrictive pattern is still unknown as no systematic pathological studies or studies using computed tomographic (CT) scanning have been performed.
We report a patient with TM who presented with severe PH, markedly increased pulmonary vascular resistance (PVR), right ventricular failure, preserved left ventricular function, a severe restrictive pattern in pulmonary function tests, and interstitial fibrosis revealed by high resolution CT (HRCT) scanning of the lung.
Case report
A 29 year old man with TM who had never smoked was admitted to the intensive care unit (ICU) with pneumonia requiring the use of a non-rebreathing mask (Fio 2 ≅ 100%) to maintain an Sao 2 of >90%. The patient had bronze coloured skin and had started blood transfusions at the age of 4, having received a total of 640 blood units to maintain haematocrit values of 26–30%. Inadequate iron chelation therapy with subcutaneous injection of desferrioxamine had been started 5 years previously (25 mg/kg/day, 2 days per week). At this time splenectomy was performed and mild systolic PH (36 mm Hg) was diagnosed by continuous wave (C-W) Doppler echocardiography through minimal tricuspid regurgitation.7 More severe systolic PH was detected 2 years later (48 mm Hg) when dyspnoea on exertion was present. PH progressed to 57 mm Hg at the age of 27; a magnetic resonance imaging (MRI) scan of the heart was within normal limits. Frequent pulmonary infections were reported during the previous 4 years.
On admission right heart failure was present (audible S3, prominent jugular veins to the angle of the jaw, pitting ankle oedema, palpable liver 15 cm below the right costal margin, and bilateral transudative pleural effusion) and digital clubbing was noted. Haematocrit was 26% (Hb 91 g/l), the platelet count was 80 000/mm3, and the serum ferritin level was 8350 ng/ml. Echocardiography showed a markedly dilated right ventricle, main pulmonary artery, and inferior vena cavae. The right ventricular free wall was hypokinetic and the interventricular septum was bulging towards the left ventricle during both systole and diastole. The left ventricle was small with preserved systolic function. Colour flow mapping disclosed moderate (2/4) tricuspid regurgitation. Systolic pulmonary artery pressure, estimated by measuring the systolic transtricuspid gradient from tricuspid regurgitation and adding it to the right atrial pressure,7 was 104 mm Hg (fig 1). The ECG was characteristic of right ventricular hypertrophy, and pulmonary scintigraphy was not compatible with pulmonary embolism. No thrombi were detected in the veins of the legs and pelvis by colour Doppler echocardiography and tests for d-dimers were negative. The patient improved quickly and was discharged from the ICU after 10 days in a stable condition, afebrile, with an Sao 2of 91% on room air.

(A) and (B) Echocardiogram on admission to ICU showing a markedly dilated right ventricle (RV) suppressing the left ventricle (LV) and moderate tricuspid regurgitation (TR). (C) and (D) Echocardiogram at recovery showing an increase in the LV to normal size and mild TR. The transtricuspid flow is barely detectable and the peak velocity has decreased from 4.7 to 4.3 m/s. (E) Mid systolic notch of pulse wave Doppler flow of pulmonary valve characteristic of severe pulmonary hypertension. The acceleration time over ejection time is markedly decreased (51/310=0.16).
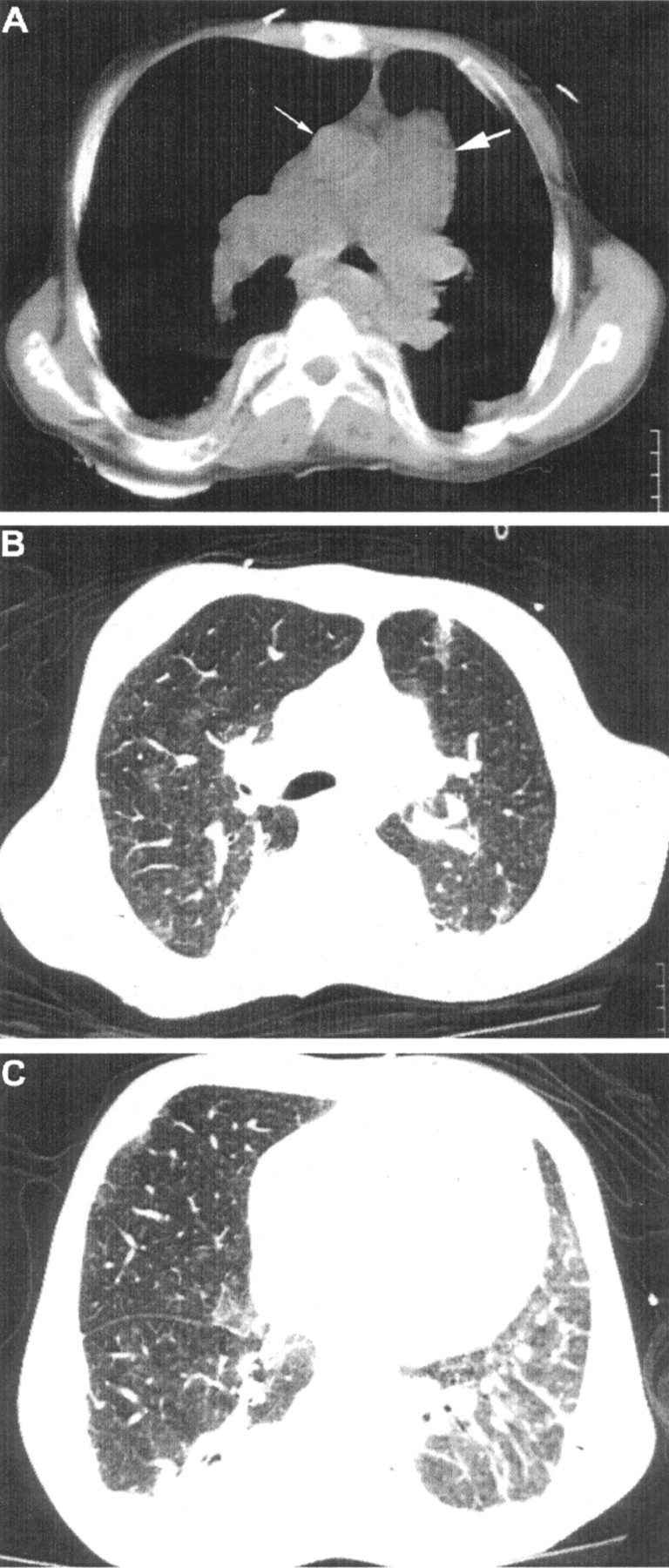
Right heart catheterisation performed 30 days later, when dyspnoea had returned to the pre-admission level (NYHA II), showed systolic and diastolic pulmonary pressures of 84 mm Hg and 47 mm Hg, respectively, and a pulmonary capillary wedge pressure (PCWP) of 12 mm Hg. PVR was 775 dynes.s/cm5 and the cardiac index, measured by thermodilution, was 2.68 l/min/m2. There was no oxygen step up in the right ventricle. An echocardiogram performed on the same day showed improvement compared with that in the ICU. Indeed, the right ventricle was less dilated, the interventricular septum was flatter, the left ventricle was of normal size, tricuspid regurgitation was mild (1/4), and systolic pulmonary pressure was estimated to be 83 mm Hg (fig 1).7 A chest HRCT scan showed severe dilatation of the main pulmonary artery and its major branches, patchy ground glass opacities in the upper lobes of both lungs, and linear and reticular interstitial thickening along the perivascular bundles of both lower lobes, mainly on the left, resulting in severe loss of volume of the left lower lobe (fig 2). Lung function tests showed a severe restrictive pattern (total lung capacity (TLC) = 3.34 l (56% predicted), residual volume (RV) = 0.88 l (54% predicted), RV/TLC = 0.26 (98% predicted), forced vital capacity (FVC) = 1.38 l (30% predicted), forced expiratory volume in 1 second (FEV1) = 1.01 l (26% predicted), FEV1/FVC 0.73 (90% predicted)) and decreased carbon monoxide transfer factor (Tlco) of 73% predicted.

{kind=link}
{kind=link}
High resolution CT scan of the chest. (A) Severe dilatation of the main pulmonary artery and its major branches. Note that the diameter of the pulmonary artery (thick arrow) is much greater than that of the ascending aorta (thin arrow). (B) Patchy ground glass opacities in the upper lobes of both lungs. (C) Linear and reticular interstitial thickening along the perivascular bundles of both lower lobes, mainly on the left. There is severe loss of volume of the left lower lobe.
Discussion
Left ventricular dilatation followed by dysfunction in the course of the disease is the usual cardiovascular finding in patients with TM.1 ,2 Right sided cardiac failure secondary to precapillary PH is quite uncommon.2 Indeed, using cardiac catheterisation, Aessopos et al found PH in only seven of a large group of patients with TM receiving regular follow up in their cardiology outpatient clinic.3 PVR ranged from 267 to 667 dynes.s/cm5. Moreover, in a population of 130 patients with TM monitored for more than 10 years, 10% of them presented with mild PH with a systolic pressure of <40 mm Hg.4 The patient we describe is therefore a rare case of severe precapillary PH with a PVR of 775 dynes.s/cm5, the highest ever reported in a patient with TM. In addition, the absence of left ventricular dysfunction assessed by echocardiography, the normal PCWP by catheterisation, and the recent normal cardiac MRI scan suggest that pulmonary impairment and PH might be responsible for right ventricular dysfunction, and not left ventricle and/or right ventricle infiltration by haemosiderosis as is more usual.2
The aetiology of precapillary PH in TM remains uncertain.3The PH seen in the patient presented here might be associated with restrictive lung disease, which is well known to occur in TM.3 ,5 ,6 However, its pathogenesis is not clear. Iron deposition in the lungs might have played a role in our patient. The high serum ferritin level and the large accumulated amount of blood transfused in association with inadequate chelation therapy, started late in life,1 were indicative of a chronic state of iron overload. Indeed, Factor et al 5have shown a strong inverse correlation between TLC and iron overload, although this has not been a universal finding.6 Iron may promote tissue damage in the lung through generation of free hydroxyl radicals or by facilitating the growth of certain microorganisms dependent on the presence of iron.8 Once damage to the parenchyma is established, it creates a suitable milieu for bacterial growth with further lung impairment.2 The recurrent pulmonary infections seen in our patient probably contributed to the generation of his restrictive pulmonary disease.
Our patient had unequivocal CT evidence of interstitial lung fibrosis. To our knowledge, this is the first time that pulmonary fibrosis has been described in a patient with TM. However, previous reports have not used CT scanning to search for interstitial fibrosis.3 ,5 ,6 Interstitial fibrosis may account for the unusually severe precapillary PH found. Other factors such as small pulmonary emboli or circulation of vasoconstrictive substances9 may also contribute to the generation of PH. In addition, pulmonary thromboembolism cannot be excluded as a contributing factor in the course of PH, although a thorough investigation proved negative.
In conclusion, interstitial lung fibrosis can complicate the course of lung involvement in patients with TM and can significantly contribute to the development of PH. The deposition and accumulation of iron over many years, as well as repeated infections, may be critical in this process in some patients.