Article Text

Abstract
BACKGROUND Nitric oxide (NO) is a product of the enzyme nitric oxide synthase (NOS) and is found in normal and asthmatic human airways. The administration ofl-arginine results in an increase in airway NO production in asthmatic subjects. This is thought to occur becausel-arginine is the substrate for NOS. However, studies in the systemic vasculature suggest that other mechanisms may be responsible.
METHODS Eight patients with steroid naive asthma each received 2.5 g l-arginine, 2.5 g d-arginine, and 2.0% saline by ultrasonic nebuliser on separate days in a randomised, single blind manner. Exhaled NO was measured by chemiluminescence and spirometric tests were performed before and for 3 hours after each administration. The mean concentration of NO after exposure was calculated from the area under the curve.
RESULTS l-arginine,d-arginine, and 2.0% saline induced a mean (95% CI) maximal bronchoconstriction of 11.9% (–1.7 to 25.4), 10.0% (2.8 to 17.2), and 8.5% (–2.5 to 19.5) of the starting forced expiratory volume in one second (FEV1), respectively. Exhaled NO declined in proportion to the degree of bronchoconstriction (r=0.60, p<0.01). Bronchoconstriction and the acute reduction in exhaled NO resolved within 15 minutes. The mean post-exposure concentration of NO was 15.75 parts per billion (ppb) after l-arginine, 15.16 ppb after d-arginine, and 12.74 ppb after 2.0% saline. The mean (95% CI) difference between l-arginine and placebo was 3.01 (0.32 to 5.7) ppb, between d-arginine and placebo 2.42 (0.10 to 4.74) ppb, and between l- and d-arginine 0.59 (–1.56 to 2.74) ppb.
CONCLUSIONS Exhaled NO decreased with acute bronchoconstriction and returned to baseline with the resolution of bronchoconstriction. Exhaled NO increased following the administration of both l-arginine andd-arginine. Since NOS is stereospecific, this finding suggests that the increase in exhaled NO is not entirely mediated through an increase in NOS enzyme activity. We suggest that arginine may react in a non-stereospecific fashion with reactive oxygen species present in asthmatic airways.
- nitric oxide
- arginine
- asthma
Statistics from Altmetric.com
Nitric oxide (NO) is found in increased amounts in the exhaled breath of patients with asthma and has been implicated in the pathophysiology of the disease.1 ,2 NO is a gaseous product of the enzyme nitric oxide synthase (NOS), of which there are both constitutive (nNOS and eNOS) and inducible (iNOS) isoforms. The substrate for NOS is the amino acid l-arginine, and the enzyme is stereoisomer specific, suggesting thatd-arginine will not act as a substrate for NOS.
The administration, either by nebulisation or orally, ofl-arginine induces an increase in NO production in the respiratory tracts of normal and asthmatic subjects.3 ,4 l-arginine also prevents methacholine induced bronchoconstriction in isolated guinea pig tracheal segments through an NO dependent mechanism.5 These findings suggest that, in both normal and asthmatic airways, there is substrate limitation for NOS.
Nitric oxide has been much better studied in vascular tissue where it was originally described as endothelium derived relaxing factor (EDRF) whose main function is to dilate arteriolar smooth muscle. In the systemic vasculature l-arginine increased NO production by endothelial cells,6 an unexpected finding since the normal intracellular concentration of l-arginine in endothelial cells is well above the Km for NOS.7 ,8 One possible explanation is that NO may be formed from l-arginine non-enzymatically through reaction with reactive oxygen species such as hydrogen peroxide.9 Since asthma is associated with oxidative stress and the formation of reactive oxygen species,10 ,11 it is possible that the increase in respiratory tract NO production after inhalation ofl-arginine in asthmatic subjects may be at least partially mediated through a non-enzymatic route. If this is the case, then inhalation of d-arginine should lead to an increase in respiratory tract NO production similar to that observed after the inhalation of l-arginine. The aim of this study was to assess whether arginine induced increases in pulmonary NO production are stereospecific.
Methods
SUBJECTS
Eight non-smoking subjects with steroid naive asthma were studied. The diagnosis of asthma was based on the presence of circadian peak flow variability of more than 20% and/or evidence of spirometric reversibility (post-bronchodilator forced expiratory volume in one second (FEV1) more than 15% greater than pre-bronchodilator FEV1) in conjunction with appropriate symptoms (breathlessness, wheezing and cough). No subject had had an upper respiratory tract infection in the 4 weeks prior to the study. β agonist medications were withheld on the morning of each visit. Subjects had not been exposed to inhaled steroid medication or to other glucocorticoids for at least 2 months before enrolment.
STUDY DESIGN
The study was of a randomised, single blind, crossover design and was approved by the East Birmingham local research and ethics committee. Each subject attended on three separate days, each at least 3 days apart. The concentration of exhaled NO was measured (mean of five measurements within 10%) and spirometric tests were performed (best of three forced exhalations within 5%) (Vitalograph-Compact, Vitalograph Ltd, Buckingham, UK) at baseline. The spirometric tests were always performed after measurement of exhaled NO. The subjects then received either l-arginine (2.5 g in 20 ml H2O, 700 mOsm, pH 6.1), d-arginine (2.5 g in 20 ml H2O, 700 mOsm, pH 6.1), or 2.0% saline control (25 ml, 684 mOsm, pH 6.5 ) nebulised ultrasonically (Intersurgical, Wokingham, UK, output approximately 4 ml/min). l- andd-arginine were supplied by Clinalfa AG, Switzerland and 2.0% saline by Martindale Pharmaceuticals, Essex, UK. The concentration of exhaled NO was measured (mean of three measurements within 10%) and spirometric tests were performed immediately after nebulisation and then again at 5, 10, 15, 30, 45, 60, 90, 120, 150 and 180 minutes after nebulisation. A single administration of 100 μg salbutamol by inhalation was allowed if the initial post-nebuliser FEV1 revealed a significant (>10%) bronchoconstriction.
ASSESSMENT OF EXHALED NO
Exhaled NO was measured by chemiluminescence (LR2000, Logan Research, Kent, UK) in accordance with European Respiratory Society guidelines12 as previously described.13Briefly, subjects inhaled to total lung capacity and then completed a slow vital capacity exhalation through a resistance with a flow meter in series. A visual feedback display allowed the subject to maintain a flow rate of approximately 200 ml/s during the exhalation, while the resistance maintained closure of the soft palate. Nose clips were not used. The chemiluminescence analyser sampled the exhalate in real time at 250 ml/min (4.2 ml/s) with a sensitivity of 0.3 parts per billion (ppb) and a sampling rate of 15 Hz. Calibration was performed daily. The exhaled NO measurement was obtained from the plateau phase at 75% of exhaled volume.13
STATISTICAL ANALYSIS
Exhaled NO and FEV1 (as % of baseline) are presented graphically as the mean at each time point for each visit. The change in FEV1 immediately following exposure was tested for statistical significance using a paired ttest. In order for comparisons to be made between exposure groups, the serial measures were summarised for each individual for each exposure from baseline to 180 minutes after exposure using the area under the curve method14 and expressed as an absolute amount by dividing by total time. Statistical significance was tested using paired t tests. A potential correlation between change in exhaled NO concentration and change in FEV1 within each subject was sought using multiple linear regression and dummy variables according to the method described by Bland and Altman.15
Results
Demographic and lung function data for the eight subjects are shown in table 1. Subjects who required salbutamol to relieve acute bronchoconstriction required it for all three visits. Immediately after exposure, l-arginine, d-arginine, and 2.0% saline induced a mean (95% CI) bronchoconstriction of 11.9% (–1.7 to 25.4, p=0.08), 10.0% (2.8 to 17.2, p<0.01), and 8.5% (–2.5 to 19.5, p=0.17) of the starting FEV1, respectively, although the degree of induced bronchoconstriction varied widely (from a 2% increase in FEV1 to a 38% decrease). These decrements in FEV1 were associated with a reduction in exhaled NO (figs 1and 2) which correlated well with the reduction in FEV1(fig 3, r=0.60, p<0.01). In all groups bronchoconstriction resolved within 15 minutes (fig 1) and exhaled NO returned to pre-exposure levels with the resolution of bronchoconstriction (fig 2). There were no differences in mean post-exposure FEV1 between the l-arginine,d-arginine, and 2.0% saline groups.
Demographic details for eight subjects with steroid naive asthma
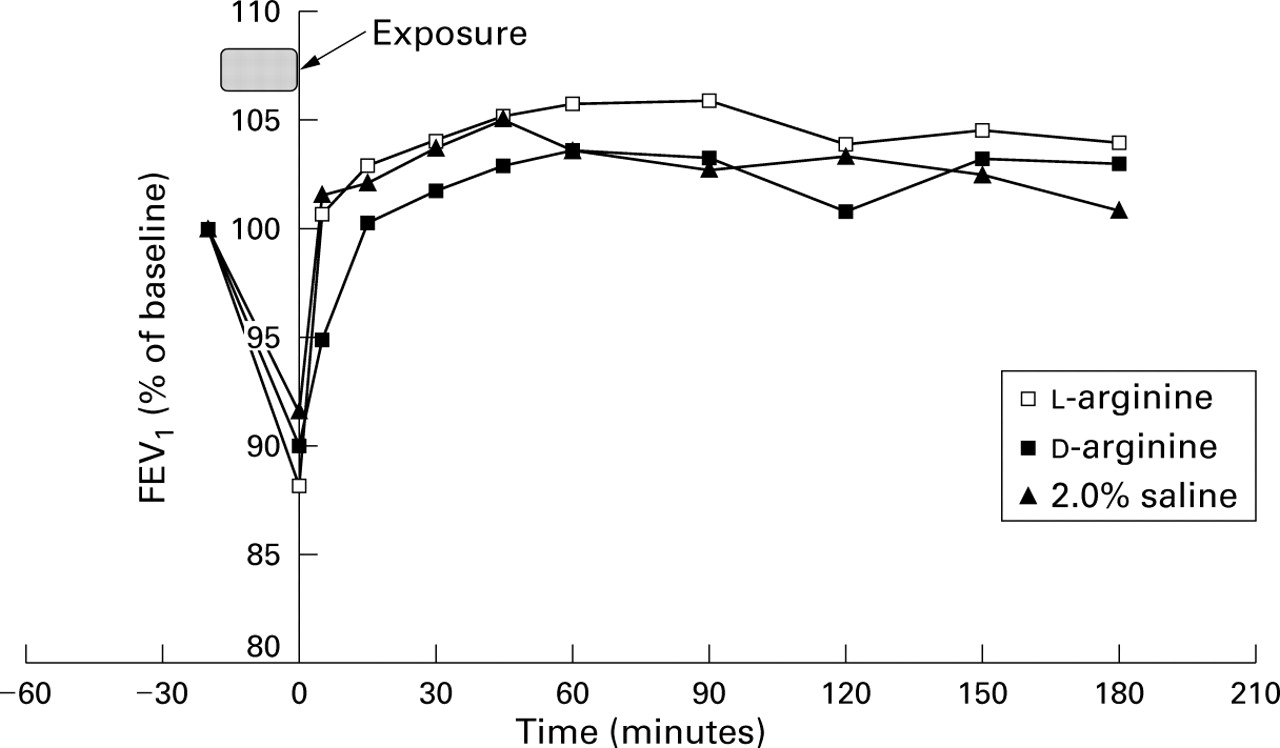
Effect of l- and d-arginine (2.5 g) and 2.0% saline inhalation on FEV1 in patients with steroid naive asthma (n=8).
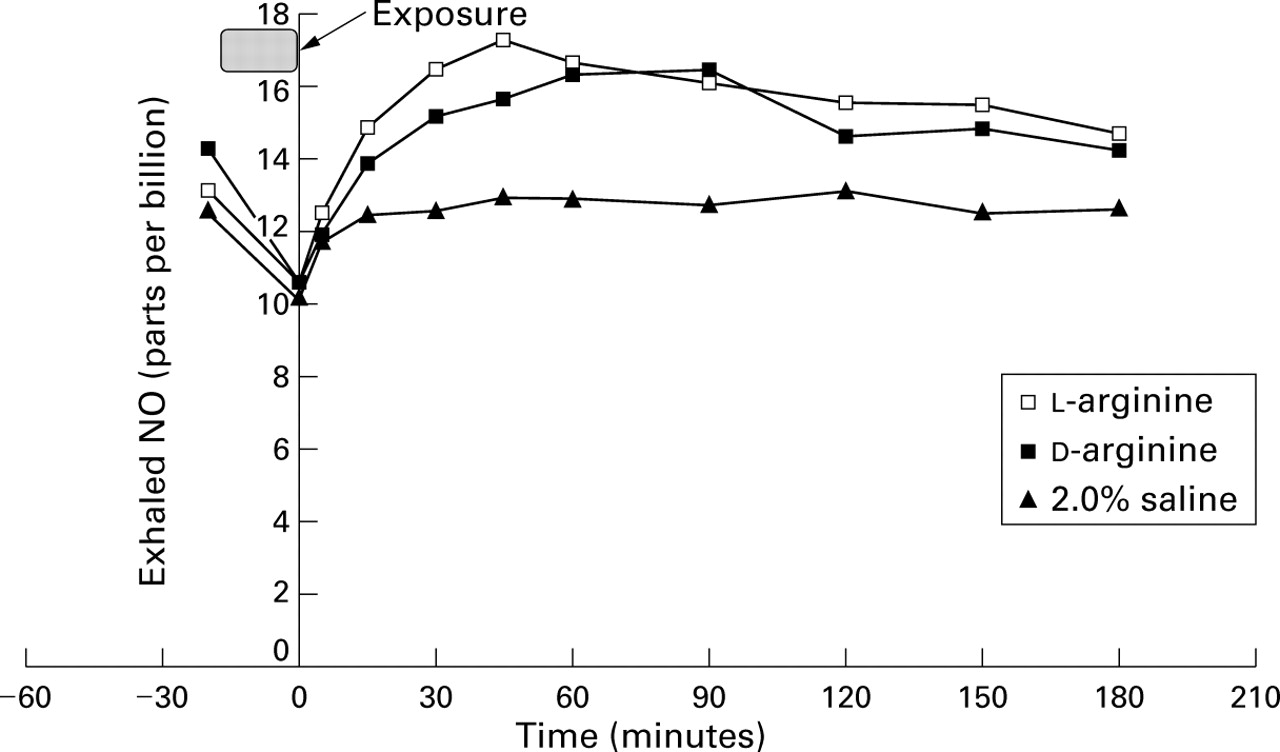
Effect of l- and d-arginine (2.5 g) and 2.0% saline inhalation on exhaled NO in patients with steroid naive asthma (n=8).

{kind=link}
{kind=link}
{kind=link}
Within subject relationship between FEV1and exhaled NO during acute bronchoconstriction (n=8, r=0.60, p<0.01).
The mean post-exposure concentrations of NO were 15.75 ppb, 16.16 ppb, and 12.74 ppb in the l-arginine,d-arginine, and 2.0% saline groups, respectively. For bothl-arginine and d-arginine the mean post-exposure concentration of NO was significantly different from placebo (p<0.05 in each case). There was no difference in post-exposure NO concentration between l- andd-arginine (p=0.54). The mean (95% CI) difference betweenl-arginine and placebo was 3.01 (0.32 to 5.70) ppb, between d-arginine and placebo 2.42 (0.10 to 4.74) ppb, and between l- and d-arginine 0.59 (–1.56 to 2.74) ppb.
Discussion
Several lines of evidence support the idea that NO is important in the pathophysiology of asthma. Firstly, it has been well shown that NO concentrations are increased in the exhaled breath of patients with asthma.1 ,2 Secondly, in patients with asthma, exhaled NO concentrations correlate with markers of airway hyperresponsiveness.16-19 Thirdly, inhibition of NO production has increased airway hyperresponsiveness in guinea pig20-22 and human airways.23 Factors which alter NO metabolism in the airways are therefore of potential significance in asthma. Most work has centred around factors which alter NOS activity or induction as these are thought to be most important in controlling NO metabolism. In this study we have studied effects on NO metabolism independent of enzyme activity or induction.
We have shown that exhaled NO increases after the administration by nebulisation of both l- and d-arginine relative to 2.0% saline control in subjects with steroid naive asthma. Additionally, acute bronchoconstriction is associated with a reduction in exhaled NO which closely parallels the reduction in FEV1. Exhaled NO concentrations return to baseline with the resolution of bronchoconstriction.
The time course and magnitude of the change in respiratory tract NO production after the administration of 2.5 g l-arginine is in good agreement with that reported by Sapienza et al after the administration of 3 g l-arginine, also by ultrasonic nebulisation.4 l-arginine may increase NO production through increased provision of substrate to NOS, as suggested by Sapienza et al. 4 However, studies in the systemic vasculature, where NO was originally described as endothelium derived relaxing factor, cast doubt on this theory. Although the administration ofl-arginine is able to increase NO production in the systemic vasculature in an analogous fashion to that reported by Sapienza et al 4 and to our findings in the lung, the intracellular concentration ofl-arginine in endothelial cells is several orders of magnitude higher than the Km for NOS,24 so it makes sense on theoretical grounds that increased substrate provision should not influence enzyme activity. Although intracellular concentrations ofl-arginine have, to our knowledge, not been measured in asthmatic airways, Sapienza et al speculate that increased NOS activity in these inflamed airways leads to a relative deficiency of l-arginine so that concentrations are low enough to be able to demonstrate substrate dependence of enzyme activity.4 However, they also found an increase in NO production after the administration of l-arginine in subjects with normal airways.4
There are a number of possible alternative explanations for these findings. Increased production of NO in the respiratory tract may be a protective response to the acute bronchoconstriction induced immediately after exposure, since NO may play a role as a bronchodilator.17 Against this theory, however, is the absence of an increase in NO production after the bronchoconstriction induced by exposure to 2.0% saline. Alternatively, l- andd-arginine may act as non-specific irritants which induce an inflammatory response in the airways, including upregulation or induction of NOS. However, NOS induction occurs over a period of hours after exposure to the irritant25 and a similar response may be expected after the administration of 2.0% saline.
Nagase and coworkers have recently described a novel non-enzymatic pathway for the generation of NO through the reaction of hydrogen peroxide with arginine.9 This reaction is not stereospecific and has been proposed as a possible explanation for the substrate dependence of NOS activity in the systemic vasculature. The concentrations of arginine and hydrogen peroxide required for the spontaneous formation of NO are in the millimolar range. Currently, the only estimates of the concentration of hydrogen peroxide in asthmatic airways come from work with breath condensates. Jobsiset al 26 found a median concentration of hydrogen peroxide of 0.6 μmol in the breath condensate of patients with stable asthma. Although this is lower than that required for the spontaneous formation of NO, it may be that higher concentrations of hydrogen peroxide are reached locally in inflamed asthmatic airways. The large doses of arginine administered in this study and the study of Sapienza et al 4 may lead to sufficiently high local airway concentrations of arginine for this reaction to become a significant source of NO.
Hunt et al 27 have recently shown that asthmatic airways are acidic during acute asthma and that this acidity resolves with treatment. They speculate that local airway chemistry may play a much more important role in the pathogenesis of asthma than previously thought. They also suggest that changes in airway chemistry are responsible for the increased pulmonary production of NO in asthma.27 ,28 Given that the pH of the saline control in this study was 6.5 while that of l- andd-arginine was 6.1, it is possible that airway acidification may have contributed to the increase in exhaled NO in the arginine groups. Against this suggestion is the finding that the administration of l-arginine orally in a dose of 0.1 and 0.2 mg/kg also increases exhaled NO.3 It is difficult to imagine an oral dose significantly altering airway pH. In addition, the pH found in exhaled breath condensate by Hunt and colleagues was very much lower and it is far from certain whether the change in pH found in acute asthma was the cause of, or associated with, an exacerbation of asthma. Regardless of the mechanism, in demonstrating that alterations in NOS activity and induction may not be entirely responsible for the alterations in NO production observed in asthma, our findings are complementary to those of Hunt et al.27
We have shown that exhaled NO concentrations decrease in parallel with decrements in FEV1 during acute bronchoconstriction and return to baseline with the resolution of bronchoconstriction. In our study, bronchoconstriction was inadvertently induced by the administration by nebulisation of hypertonic solutions ofl-arginine, d-arginine, and saline. A decrease in exhaled NO concurrent with acute bronchoconstriction and its resolution with bronchodilation has been well described previously29-31 but the mechanism remains unknown. In particular, it is not known whether the decrease in exhaled NO is causal or merely associated with bronchoconstriction. The simplest explanation would be that the bronchial epithelial surface area declines with bronchoconstriction, so reducing the available area from which NO may diffuse toward the lumen. However, Jameset al have shown that the bronchial mucosa concertinas with bronchoconstriction so that the epithelial surface area remains constant.32 Alternatively, the thickness of the epithelial lining fluid (ELF) may increase with bronchoconstriction as airway diameter decreases. Thickening and increased tenacity of the ELF is thought to be the mechanism for the reduction in exhaled NO in patients with cystic fibrosis.33 Another explanation which remains completely unexplored is that the decrease in exhaled NO is a primary event and gives rise to bronchoconstriction. Further work in this area will provide further insight into the pulmonary physiology of NO.
In summary, we describe an increase in the concentration of exhaled NO after the administration of both l- andd-arginine relative to 2.0% saline control in subjects with steroid naive asthma, and acute decreases in exhaled NO concurrent with and in proportion to bronchoconstriction. The increase in exhaled NO after d-arginine suggests that this effect is not entirely mediated through NOS. We suggest that arginine may react directly with hydrogen peroxide present in asthmatic airways to form NO and that changes in airway chemistry (for instance redox state) may modulate NO release with potential pathophysiological consequences.
References
Narrative Based Medicine, An Interdisciplinary Conference Research, Narrative, and Practice A two day conference—Monday 3rd and Tuesday 4th September 2001 Homerton College, Cambridge, UK
BMJ Publishing Group
For full details contact: BMA/BMJ Conference Unit, Tavistock Square, London, WC1H 9JP Tel: +44 (0)20 7383 6819; fax: +44 (0)20 7383 6663; email: clyders{at}bma.org.uk. www.quality.bmjpg.com