Article Text

Abstract
BACKGROUND Transforming growth factor β1 is implicated in the pathogenesis of lung fibrosis. It promotes extracellular matrix accumulation by increasing procollagen synthesis and reducing degradation. TGFβ1 gene and protein expression increase in experimental lung fibrosis, and TGFβ1 antibodies attenuate fibrosis in mice. The role of other TGFβ isoforms is unclear. This study aimed to localise TGFβ1 and TGFβ3 gene expression in fibrotic human lung and compare it with that in normal human lung.
METHODS Lung tissue from patients with cryptogenic fibrosing alveolitis and fibrosis associated with systemic sclerosis was examined by in situ hybridisation. Macroscopically normal lung from carcinoma resections was used as control tissue. Digoxigenin labelled riboprobes were synthesised from TGFβ isoform specific cDNA templates.
RESULTS The digoxigenin labelled riboprobes were sensitive and permitted precise cellular localisation of mRNA transcripts. TGFβ1 and TGFβ3 mRNA transcripts were widespread in normal lung and localised to alveolar macrophages and bronchiolar epithelium. TGFβ1 but not TGFβ3 mRNA was detected in mesenchymal and endothelial cells. In fibrotic lung tissue mRNA transcripts for both isoforms were also detected in metaplastic type II cells. TGFβ1 gene expression was enhanced in some patients. TGFβ3 was expressed in fibrotic lung but was not consistently altered compared with controls.
CONCLUSION TGFβ1mRNA transcripts were localised in normal and fibrotic human lung and TGFβ3 gene expression in human lung fibrosis was shown for the first time. The results suggest that TGFβ1 may play the predominant role in pathogenesis. It is suggested that TGFβ1 should be the primary target of anticytokine treatments for pulmonary fibrosis.
- pulmonary fibrosis
- transforming growth factor β
- gene expression
Statistics from Altmetric.com
The pathogenesis of pulmonary fibrosis is not yet fully elucidated but one current hypothesis is that cytokines released by resident cells and inflammatory cells entering the lung stimulate fibroblasts to synthesise excessive amounts of extracellular matrix including collagen.1 The transforming growth factor beta (TGFβ) family has been implicated in the pathogenesis. TGFβ1 is a potent promoter of extracellular matrix deposition and acts by increasing production as well as reducing degradation.2 TGFβ1 gene expression and protein synthesis increase in experimental3 ,4 and human pulmonary fibrosis.5 ,6 Enhanced pulmonary expression of active TGFβ1 induces a chronic fibrotic response,7 while serum TGFβ1 levels predict pneumonitis and lung fibrosis in patients undergoing radiotherapy.8 ,9 Furthermore, the homozygous TGFβ1 genotype, which correlates with excessive TGFβ1 expression, is a risk factor for allograft lung fibrosis.10
The role of the other mammalian TGFβ isoforms, TGFβ2and TGFβ3, remains unclear. We have previously shown that all three isoforms stimulate synthesis of human fibroblast procollagen and reduce intracellular degradation in vitro. We have also demonstrated differential TGFβ isoform gene expression in murine bleomycin induced lung fibrosis.11 The data suggest that, unlike TGFβ1, TGFβ3 is not involved in the pathogenesis. Finally, we have shown that TGFβ1 and TGFβ3 mRNA transcripts are widespread in normal murine and human lung.12 The aims of this present study were to determine whether TGFβ3 is expressed in fibrotic human lung and to compare the localisation of TGFβ1 and TGFβ3 mRNA transcripts in fibrotic human lung with that in normal human lung.
Methods
TISSUE PREPARATION
Lung tissue was obtained from open lung biopsy specimens of eight patients with pulmonary fibrosis, five from patients with pulmonary fibrosis associated with systemic sclerosis (SSc) and three from patients with cryptogenic fibrosing alveolitis (CFA). Patients with CFA were diagnosed following history and examination, investigations including high resolution computed tomography and pulmonary function testing, and exclusion of other risk factors for pulmonary fibrosis such as dust exposure or connective tissue disease. All patients with SSc fulfilled the American Rheumatism Association preliminary criteria for the diagnosis of systemic sclerosis. No patients were receiving treatment for lung disease at the time of investigation or biopsy. Macroscopically normal lung from two patients undergoing resection for lung cancer was used as a control. Only macroscopically and histologically normal peripheral lung was chosen for these studies. Viable tissue was fixed in freshly prepared 4% paraformaldehyde in phosphate buffered saline (PBS). After 4 hours immersion in fixative, lung tissue was transferred to 15% sucrose in PBS prior to dehydration and embedding in paraffin wax.
Prehybridisation treatments were performed using techniques described previously.12 Briefly, 5 μm sections were cut and placed on slides previously coated with a 2% v/v solution of 3-aminopropyltriethoxysilane in acetone. After dewaxing, sections were rehydrated through a series of ethanol washes of decreasing concentration followed by immersion in 0.14 mol/l sodium chloride and PBS before refixing in freshly prepared 4% paraformaldehyde. In order to maximise probe penetration into cells, sections were treated with proteinase K (Life Technologies, Paisley, UK) at a concentration of 20 μg/ml in 50 mmol/l Tris hydrochloride, pH 7.5, 5 mmol EDTA for 10 minutes before refixing with paraformaldehyde. Sections were then acetylated by immersion in freshly prepared 0.1 mol/l triethanolamine containing 0.25% acetic anhydride and subsequently dehydrated through a series of increasing concentrations of ethanol.
PROBE PREPARATION
Riboprobes were synthesised from transcript specific murine TGFβ cDNA constructs in pGEM vectors. The constructs were obtained by deletion of the highly conserved regions of the two murine cDNAs and the specificity of riboprobes synthesised from these templates is established.13 ,14 The TGFβ1 construct includes nucleotides 421–1395 of the murine cDNA and contains 764 bp of the amino terminal glycopeptide (precursor) region and 210 bp of the mature region. The TGFβ3 construct contains 609 bp of the amino terminal glycopeptide region (831–1440). The TGFβ isoforms are well conserved between species at peptide, N-terminal precursor region, and genomic levels.15 ,16
Digoxigenin labelled riboprobes were synthesised by in vitro transcription from the linearised cDNA templates using the appropriate RNA polymerase (SP6 or T7) according to the manufacturer's protocol (Boehringer Mannheim, Lewes, UK).
IN SITU HYBRIDISATION
This protocol was based on previously described methods.11 ,12 Hybridisation buffer consisting of 50% deionised formamide, 300 mmol /l NaCl, 20 mmol/l Tris HCl (pH 7.4), 5 mmol/l EDTA, 10 mmol monosodium phosphate (pH 8.0), 10% dextran sulphate, 1 × Denhardt's solution, and 500 μg/ml yeast tRNA was mixed with the digoxigenin labelled probe at a ratio of 9:1 to give a final probe concentration of 20 ng/ml. A 25 μl aliquot of hybridisation solution was applied to each slide and sections were incubated overnight (16 hours) at 50°C in a sealed chamber humidified with a solution of 50% formamide in 2 × saline sodium citrate (SSC). After hybridisation, all incubations were performed at room temperature. Slides were washed in 4 × SSC for 30 minutes, then in 0.2 × SSC for 30 minutes, and finally in Tris buffered saline (TBS, consisting of 0.1 mol/l Tris, pH 8.2, 0.15 mol/l NaCl) for 5 minutes. They were then incubated for 30 minutes with antibody blocking solution consisting of 5% bovine serum albumin and 5% normal sheep serum diluted in TBS with 0.1% Tween 20 (Sigma, Dorset, UK). After two additional 5 minute washes in TBS, the slides were incubated with antibody solution for 30 minutes. This consisted of a 1:100 dilution of anti-digoxigenin-alkaline phosphatase Fab fragments (Boehringer Mannheim) in 1% bovine serum albumin in TBS with Tween 20. Sections were then washed twice in TBS for 5 minutes each.
For detection of bound antibody, sections were incubated with alkaline phosphatase substrate New Fuschin Red (Dako, High Wycombe, UK). This was prepared according to the manufacturer's instructions and 1 mmol/l levamisole was added to inhibit endogenous alkaline phosphatase activity.17 A 200 μl aliquot of freshly prepared reagent was applied to each section for 20 minutes. The slides were then rinsed in distilled water and counterstained with haematoxylin or methyl green. New Fuschin Red yields a red colour at the site of hybridised probe. Each in situ experiment was repeated at least once.
Sections were examined and reported independently by three of the authors (RKC, PKJ, RJM). Control and fibrotic sections were tested in the same hybridisation run. The cell types expressing TGFβ were compared and the intensity of signal rated on a scale of mild (+), moderate (++) or intense (+++). Expression in fibrotic lung was compared with expression in control sections. The slides were blinded and the results discussed. Where agreement could not be reached as to whether signal was increased, unchanged or decreased, these slides were excluded from the analysis.
Results
PATIENT CHARACTERISTICS
Control sections were analysed from two patients, a woman aged 59 years with bronchial adenocarcinoma and a man aged 44 years with mixed small cell carcinoma and adenocarcinoma. Both were smokers. Fibrotic specimens were examined from five patients with SSc and three with CFA. The patients with SSc were women ranging in age from 36 to 55. Two of the patients with CFA were men aged 64 and 66 and the third was a woman aged 42. Three patients with SSc had a predominantly cellular pattern and two had a predominantly fibrotic pattern of disease. Two patients with CFA had advanced fibrosis and one had patchy fibrosis.
TGFβ ISOFORM EXPRESSION IN NORMAL HUMAN LUNG
As reported previously,12 TGFβ1 and TGFβ3 mRNA transcripts were widespread in normal human lung and localised predominantly to alveolar macrophages and bronchiolar epithelium. Figure 1a, c, and d illustrate the typical cellular localisation of TGFβ1 gene expression in normal human lung; TGFβ1 mRNA transcripts were localised to bronchial epithelium (arrows in fig 1a), alveolar macrophages (arrows in fig 1c), alveolar walls (arrowheads in fig 1c), and mesenchymal cells (arrows in fig 1d). Figure 1e, g, and h illustrate the typical cellular localisation of TGFβ3 gene expression in normal human lung; TGFβ3 mRNA transcripts were localised to alveolar macrophages (arrows in fig 1e and g) and bronchial epithelium (arrows in fig 1h). No hybridisation signal for TGFβ1 or TGFβ3 was detected with sense riboprobes (fig 1b and f, respectively). TGFβ1 but not TGFβ3 mRNA transcripts were also detected in pulmonary endothelial cells (data not illustrated).

In situ hybridisation in normal human lung with TGFβ1 and TGFβ3 riboprobes. Hybridisation signal for TGFβ1 was evident in bronchial epithelium (a), alveolar macrophages (arrows in c), alveolar walls (arrowheads in c), and mesenchymal cells (d). TGFβ3 mRNA transcripts were localised to alveolar macrophages (e and g) and bronchial epithelium (h). No hybridisation signal was obtained with TGFβ1 or TGFβ3 sense riboprobes (b and f, respectively). Counterstain in a, b, d, e, and g: methyl green. Internal scale bar = 50 μm; original magnification variable from ×200 to ×1000.
TGFβ ISOFORM EXPRESSION IN FIBROTIC HUMAN LUNG
In fibrotic lung tissue mRNA transcripts for both isoforms were also localised to alveolar macrophages, bronchial epithelial cells, alveolar epithelial cells, and mesothelial cells. TGFβ1mRNA transcripts were furthermore detected in mesenchymal cells. Gene expression for neither isoform was detected in pulmonary endothelial cells in fibrotic lung tissue. Figure 2 illustrates the typical cellular localisation of TGFβ1 and TGFβ3gene expression in fibrotic human lung. TGFβ1 mRNA transcripts were localised to areas of inflammatory cell infiltrate (fig 2a) and bronchial epithelium (fig 2b) in a patient with SSc, and to alveolar macrophages in a patient with CFA (fig 2c). TGFβ1 mRNA transcripts were also localised to type I alveolar epithelial cells (fig 2d) and mesothelial cells (fig 2e) in a patient with SSc. TGFβ3 mRNA transcripts were localised to inflammatory cells (fig 2h), bronchial epithelium (fig 2i), and mesothelial cells (fig 2j) in a patient with SSc. Hybridisation signal for TGFβ3 was also noted in intra-alveolar macrophages (arrowheads in fig 2k) and in cuboidal type I alveolar epithelial cells lining the alveolar surface (arrows in fig 2k). This lung tissue was taken from a patient with SSc. No hybridisation signal for TGFβ1 or TGFβ3 was detected using sense riboprobes (panels fig 2f and g, respectively).
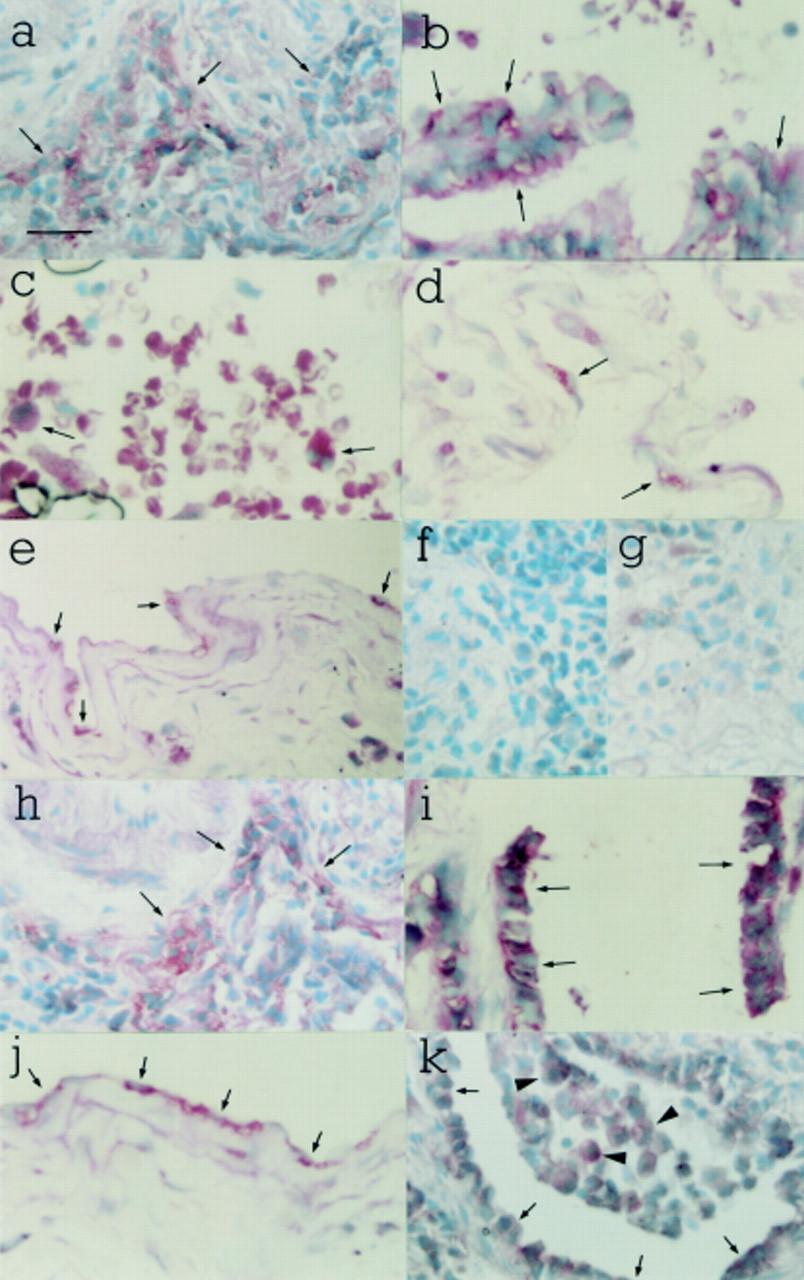
In situ hybridisation in fibrotic human lung with TGFβ1 and TGFβ3 riboprobes. TGFβ1 mRNA transcripts were localised to inflammatory cells (a) and bronchial epithelium (b) in a patient with SSc, and to alveolar macrophages in a patient with CFA (c). TGFβ1mRNA transcripts were also localised to type I alveolar epithelial cells (d) and mesothelial cells (e) in a patient with SSc. TGFβ3 mRNA transcripts were localised to inflammatory cells (h), bronchial epithelium (i) and mesothelial cells (j) in a patient with SSc. Hybridisation signal for TGFβ3 was also noted in intra-alveolar macrophages (arrowheads in k) and in cuboidal type I alveolar epithelial cells lining the alveolar surface (arrows in k). This lung tissue was taken from a patient with SSc. No hybridisation signal was obtained with TGFβ1 or TGFβ3 sense riboprobes (f and g, respectively). Counterstain: methyl green. Internal scale bar = 50 μm; original magnification ×400.
Figure 3 illustrates the absence of TGFβ1 and TGFβ3 gene expression in advanced lung fibrosis. Consecutive sections of lung tissue from a patient with CFA and advanced fibrosis were hybridised with the TGFβ1 (fig 3a) and TGFβ3 (fig 3b) antisense riboprobes. No hybridisation signal was detected for either isoform in sections examined from this biopsy specimen.

{kind=link}
{kind=link}
{kind=link}
Lung tissue from a patient with CFA and advanced fibrosis hybridised with (a) TGFβ1 and (b) TGFβ3 antisense riboprobes showing the absence of TGFβ1 and TGFβ3 gene expression in advanced lung fibrosis. No hybridisation signal for either isoform was detected in sections. Counterstain: methyl green. Internal scale bar = 50 μm; original magnification ×400.
The cells expressing TGFβ1 and TGFβ3 in normal and fibrotic human lung are summarised in table 1. There was no difference in localisation between pulmonary fibrosis associated with SSc and CFA.
Cell types expressing TGFβ1 and TGFβ3
SEMI-QUANTITATIVE ANALYSIS OF CHANGES IN TGFβ1 AND TGFβ3 GENE EXPRESSION IN FIBROTIC HUMAN LUNG
Semi-quantitative changes in TGFβ isoform gene expression in patients with CFA and SSc are shown in table 2. TGFβ1gene expression was increased in three of the eight biopsy specimens while TGFβ3 gene expression was enhanced in only one.
Quantitative changes in TGFβ1 and TGFβ3 gene expression in fibrotic human lung
Discussion
TGFβ1 AND TGFβ3 EXPRESSION IN NORMAL LUNG
Few studies to date have examined TGFβ1 gene expression in normal adult human lung. TGFβ1 mRNA transcripts have been localised to alveolar macrophages6 ,18 using isotopic in situ hybridisation, and Northern analysis has demonstrated TGFβ1 gene expression in the airways of healthy smokers.19 We have previously reported both TGFβ1 and TGFβ3 gene expression in adult human lung,12 the findings of which are confirmed by this present study.
The data presented here show that mRNA transcripts for both TGFβ1 and TGFβ3 are present in normal human lung and predominantly co-localised to bronchiolar epithelium and alveolar macrophages. TGFβ1 gene expression was also observed in pulmonary endothelial and mesenchymal cells. Immunohistochemical studies in normal human lung and healthy smokers have demonstrated TGFβ in bronchial epithelium, alveolar macrophages, smooth muscle cells, and blood vessels,19 ,20 but some workers report little or no TGFβ1 in normal lung.21 ,22 The latter study demonstrated TGFβ3 protein in alveolar macrophages, epithelial, and smooth muscle cells in normal human lung. Taken together, these data suggest that, as we have previously shown for murine lung,12 gene and protein expression for TGFβ1 and TGFβ3 are co-localised, indicating an autocrine as well as paracrine mode of action. As in murine lung, the diversity of cells expressing these genes is consistent with the wide variety of biological functions ascribed to these peptides.
TGFβ1 AND TGFβ3 EXPRESSION IN FIBROTIC LUNG
TGFβ1 gene expression appeared to be increased in three of the eight patients while TGFβ3 gene expression was increased in only one. In patients with dense fibrosis there was a paucity of hybridisation signal for either isoform. These results confirm the findings in murine bleomycin induced lung fibrosis that the isoforms are generally co-localised but that differential gene regulation occurs. Although the numbers of patients are too small for definitive conclusions to be drawn, there was a tendency for TGFβ1 to be the predominant isoform expressed in fibrotic lung. Since TGFβ1 gene and protein expression23 and serum levels24 are increased in lung cancer, and our control lung tissue was obtained from tumour resections, the increases observed in TGFβ1 and TGFβ3 gene expression may be an underestimate.
Human lung tissue was taken from current or previous smokers, raising the possibility that smoking might alter TGFβ gene expression. To date no studies have addressed this question directly. Although nicotine reduces TGFβ1 release by bovine aortic endothelial cells in vitro,25 it does not alter TGFβ1 gene expression. A study of TGFβ1gene expression in human airways showed no differences between smokers and non-smokers.19
Our findings suggest that TGFβ1 may be more consistently implicated in the pathogenesis of pulmonary fibrosis associated with SSc or CFA than is TGFβ3. This is consistent with the findings of Khalil and colleagues22 who reported ubiquitous TGFβ2 and TGFβ3 protein in normal human lung but little TGFβ1. In contrast, TGFβ1 was widespread in macrophages, epithelial cells, and extracellular matrix in fibrotic lung. TGFβ2 and TGFβ3 deposition was unchanged in fibrotic lung, suggesting that the presence of TGFβ1 is a marker of chronic fibrosis. However, since the profibrotic potential of TGFβ3 in vitro is at least equivalent to that of TGFβ1 26 and may exceed it,11the possibility that this isoform does play an important role in some patients, or at a particular stage of the disease, cannot yet be excluded.
Both TGFβ1 and TGFβ3 mRNA transcripts were observed in fibrotic lung and their distribution appeared to be identical, which suggests that gene activation occurs simultaneously. Gene expression for both isoforms was observed in alveolar macrophages, bronchial epithelium, alveolar walls, and mesothelial cells. No differences in localisation of TGFβ1 and TGFβ3 gene expression were detected between patients with CFA or SSc. This reflects the affinity between these conditions in terms of their histology and radiology, and suggests that aspects of their pathogenesis—including their profile of TGFβ induction—are similar. Similarly, a study of TGFβ1 protein deposition in fibrotic lung showed no difference between patients with idiopathic pulmonary fibrosis, asbestosis, hypersensitivity, or non-specific pneumonitis.21
The prognosis of pulmonary fibrosis associated with SSc is better than for patients with CFA.27 This may in part be because pulmonary fibrosis in SSc is generally detected earlier and in younger patients. However, enhanced TGFβ gene and protein expression, while important in the pathogenesis of the disease, may not be the sole determinant of prognosis and other factors may be implicated.
In contrast with our findings in the murine model,11 a switch from predominant TGFβ1 gene expression in bronchial epithelium to predominantly interstitial expression following lung injury was not observed in human fibrotic lung. There are several possible explanations for this. Firstly, the bleomycin model is a model of acute lung injury rather than slowly progressive disease. Changes in localisation of gene expression may therefore be more subtle and less easily detected in the human disease. Secondly, human airway epithelial cells may be subject to a greater degree of activation than those of laboratory mice, leading to higher basal TGFβ gene expression. Finally, patients with pulmonary fibrosis often present relatively late. It is therefore possible that an obvious switch does occur early on in the disease but was no longer apparent by the time the biopsy specimens were taken.
A previous study has examined TGFβ1 gene expression in human fibrotic lung.6 This study, employing a radiolabelled probe, localised TGFβ1 gene expression to macrophages adjacent to fibrotic foci but did not detect other cells expressing the gene. This supports our previous suggestion that digoxigenin labelled riboprobes are probably more sensitive tools than radiolabelled probes for detecting cytokine gene expression in lung tissue.
Immunohistochemical studies of TGFβ in pulmonary fibrosis have previously localised the protein to fibroblastic foci,6alveolar macrophages, bronchiolar epithelial and hyperplastic alveolar type II cells.5 ,21 Our data show a similar distribution for TGFβ1 and TGFβ3 mRNA. These data suggest co-localisation of gene and protein expression and therefore a predominantly autocrine and/or paracrine mode of action for TGFβ. This is consistent with previously published findings in murine lung.
Hybridisation signal for both TGFβ1 and TGFβ3 was scarce in biopsy specimens characterised by areas of dense acellular fibrosis. This may reflect the fact that the predominant source of TGFβ is inflammatory cells so that, when few of these cells are present, little gene expression is observed. This would be consistent with Broekelmann's data showing that mRNA transcripts for TGFβ1 were associated with macrophages while TGFβ1 was associated with areas of extracellular matrix deposition.6 It is also consistent with observations by Asakura and colleagues who found that TGFβ1 protein deposition is associated with early rather than late fibrosis in patients with pulmonary adenocarcinoma.28
Another possibility is that TGFβ gene expression is downregulated by matrix protein accumulation. TGFβ1promoter transcription and gene expression by mammary epithelial cells are downregulated when these cells are cultured in contact with extracellular matrix rather than on plastic.29 This phenomenon was not observed with TGFβ2 and no data are available for TGFβ3. In vivo studies have shown that TGFβ1 and decorin protein deposition are inversely related in patients with lung fibrosis and adenocarcinoma.28 These findings are consistent with the recognised ability of various extracellular matrix components to bind and sometimes inactivate active TGFβ1, including betaglycan,30 decorin,31 and type IV collagen.32 It might also provide a mechanism whereby tissue injury, involving disruption of the interaction between cells and their basement membranes, promotes enhanced TGFβ1gene expression. If this is the case, absence of TGFβ1mRNA transcripts in murine lung bronchial epithelial cells following bleomycin presumably reflects injury severe enough to inhibit gene transcription.
Finally, biopsy specimens characterised by dense fibrosis may simply represent an advanced stage of the disease where the stimulus for increased TGFβ gene expression is no longer present. If this is the case, a very low level of TGFβ gene expression is presumably sufficient to maintain increased extracellular matrix deposition. It is likely that a combination of the above factors accounts for the reduction in TGFβ gene expression observed in association with densely fibrotic lung tissue.
In summary, mRNA transcripts for both isoforms are predominantly co-localised in normal lung to bronchiolar epithelium and alveolar macrophages. TGFβ1 gene expression was also observed in pulmonary endothelial and mesenchymal cells. In fibrotic lung, gene expression for both isoforms was seen in alveolar macrophages, bronchial epithelium, alveolar walls, and mesothelial cells. No differences in localisation of TGFβ1 and TGFβ3 gene expression were detected between patients with CFA or SSc. TGFβ1 gene expression was more consistently enhanced in fibrotic lung than was TGFβ3 gene expression, which suggests that TGFβ1 may be more frequently implicated in the pathogenesis of pulmonary fibrosis. Gene expression for both isoforms was predominantly localised to areas of inflammation and mild fibrosis. Little hybridisation signal was seen in regions of dense acellular fibrosis.
These results confirm our previous reports of the high sensitivity of digoxigenin labelled riboprobes for localisation of cytokine gene expression in lung tissue. Together with previously published immunohistochemical studies they suggest autocrine and paracrine modes of action for TGFβ in human lung and support the view that TGFβ plays multiple roles in normal human pulmonary homoeostasis. As in the murine model of lung fibrosis, our results show that the isoforms can be regulated differentially, but suggest that the two different forms of pulmonary fibrosis behave similarly in terms of TGFβ gene expression.
In conclusion, we have reported TGFβ3 gene expression in fibrotic human lung tissue for the first time. In some patients with lung fibrosis TGFβ1 gene expression was enhanced. TGFβ3 was expressed in fibrotic lung but was not consistently altered compared with controls. Future anticytokine treatments for pulmonary fibrosis may need to be directed predominantly against TGFβ1.
Acknowledgments
We are grateful for the gift of cDNA probes for TGFβ1 and TGFβ3 from Professor Harold Moses (Nashville, Tennessee, USA). The work was funded by the Wellcome Trust (London, UK).
References
Narrative Based Medicine, An Interdisciplinary Conference Research, Narrative, and Practice A two day conference—Monday 3rd and Tuesday 4th September 2001 Homerton College, Cambridge, UK
BMJ Publishing Group
For full details contact: BMA/BMJ Conference Unit, Tavistock Square, London, WC1H 9JP Tel: +44 (0)20 7383 6819; fax: +44 (0)20 7383 6663; email: clyders{at}bma.org.uk. www.quality.bmjpg.com