Article Text

Abstract
BACKGROUND Nitric oxide (NO) and prostanoids are mediators of vascular and bronchial tone that are postulated to be involved in asthma. Increased levels of both are found in asthmatic subjects and are synthesised by enzymes that have cytokine inducible forms: inducible NO synthase (iNOS) and cyclo-oxygenase-2 (COX-2), respectively. We hypothesised that the in vivo expression of iNOS and COX-2 in the airways would be increased in asthma, and that these cytokine inducible enzymes may represent targets for regulation by corticosteroid treatment.
METHODS Bronchial biopsy specimens were obtained from three groups of subjects: atopic asthmatics treated with β2 agonists alone (n=7), atopic asthmatics additionally receiving regular treatment with corticosteroids (n=8), and non-asthmatic control subjects (n=10). Expression of iNOS and COX-2 mRNA and immunoreactive protein was studied using in situ hybridisation and quantitative immunohistochemistry.
RESULTS Immunoreactivity and the hybridisation signal for iNOS and COX-2 were mainly localised in the airway epithelium. The proportion of epithelium immunostained was significantly greater in the non-steroid treated asthmatic subjects (iNOS 8.6 (1.8)%; COX-2 26.3 (4.6)%) than either the steroid treated asthmatics (iNOS 3.4 (1.0)%, p=0.009; COX-2 13.0 (0.6)%, p=0.0015) or the non-asthmatic controls (iNOS 4.2 (0.9)%, p=0.018; COX-2 11.6 (0.6)%, p=0.0003). Similarly, the hybridisation signal was stronger in the non-steroid treated group of asthmatic subjects than in the other two groups.
CONCLUSIONS These findings highlight the potential role of the airway epithelium both as a contributor to the inflammatory process in asthma and as a target for inhaled corticosteroid treatment in this disease.
- asthma
- corticosteroids
- immunohistochemistry
- in situ hybridisation
- nitric oxide synthase
- cyclo-oxygenase (COX)
Statistics from Altmetric.com
- asthma
- corticosteroids
- immunohistochemistry
- in situ hybridisation
- nitric oxide synthase
- cyclo-oxygenase (COX)
Mucosal inflammation in asthma is characterised by infiltration and activation of immune cells—principally mast cells, eosinophils, T lymphocytes, and monocytes/macrophages—and activation of structural cells such as airway epithelial cells and subepithelial myofibroblasts.1 These various cell populations are believed to release biochemical mediators whose biological properties determine the clinical and pathophysiological expression of the disease. Prominent among these putative mediators are nitric oxide (NO) and prostanoids.
NO plays an important role in a wide variety of pulmonary functions including regulation of vascular and airway smooth muscle tone. Endogenous NO is normally present in the exhaled air of animals and humans.2 Levels of exhaled NO are increased in asthma3-5 and are reduced by corticosteroid treatment.6 ,7 NO is generated by the enzyme NO synthase (NOS) which catalyses the conversion of l-arginine tol-citrulline. Three major forms of NOS have been recognised, all of which are present in human airways.8 ,9Endothelial NOS (eNOS) and neuronal NOS (nNOS) are constitutively expressed and produce small (picomolar) amounts of NO that are probably involved in normal pulmonary homeostasis. In contrast, inducible NOS (iNOS) is transcriptionally regulated by cytokines and produces much larger (nanomolar) quantities of NO.
Prostanoids are also centrally involved in pulmonary physiology and have been implicated in the pathophysiology of diseases such as asthma.10 Of particular relevance is their ability to regulate bronchial tone: prostaglandin(PG)D2, PGF2α, and 9α11β-PGF2α are potent bronchoconstrictors11 ,12 whereas PGE2 is capable of causing either contraction or relaxation of airway smooth muscle.12 ,13 Levels of prostanoids in bronchoalveolar lavage (BAL) fluid are increased in asthma,14 as is release of PGE2 by cultured bronchial epithelial cells derived from asthmatic subjects.15 The initial rate limiting step in prostanoid biosynthesis is the conversion of arachidonic acid to PGH2 by the enzyme cyclo-oxygenase (COX), also known as prostaglandin endoperoxide synthase. In a variety of species two distinct but homologous COX isoenzymes, denoted COX-1 and COX-2, that are encoded by separate genes have been identified and cloned.16-18 Under most conditions COX-1 is constantly expressed and is believed to produce low levels of prostanoids which serve a physiological housekeeping role. In contrast, COX-2 is readily inducible in cell culture studies by such proinflammatory stimuli as lipopolysaccharide, interleukin (IL)-1, and tumour necrosis factor (TNF)α, and is inhibited by corticosteroids.17-19 It has been proposed that COX-2 is centrally implicated in the generation of large quantities of prostanoids during inflammatory processes.
We hypothesised that the in vivo expression of iNOS and COX-2 in the airways would be increased in asthma, and that these cytokine inducible enzymes may represent targets for regulation by corticosteroids. To investigate this we obtained bronchial biopsy specimens from three groups of subjects: atopic asthmatic subjects treated with β2 agonists alone, atopic asthmatic subjects additionally receiving regular treatment with corticosteroids, and non-asthmatic control subjects. We used in situ hybridisation and quantitative immunohistochemistry to study the expression of iNOS and COX-2 mRNA and immunoreactive protein.
Methods
SUBJECTS
Ten non-asthmatic control subjects (3M/7F, mean (SE) age 22.7 (1.5) years), seven atopic asthmatic subjects treated only with inhaled short acting β2 agonists as required (1M/6F, mean (SE) age 20.7 (0.5) years), and eight atopic asthmatic subjects who were additionally receiving regular maintenance treatment with inhaled and/or oral corticosteroids (4M/4F, mean (SE) age 29.6 (4.5) years) took part in the study. The clinical, physiological, and treatment details of the subjects in these three groups are summarised in table1. Subjects in the steroid treated group had been receiving corticosteroid therapy at the doses shown (or higher) for at least 4 weeks. The study conformed to the Declaration of Helsinki and was approved by the local ethics committees. All subjects gave written informed consent prior to participation.
Clinical and physiological characteristics of the subjects studied
PREBRONCHOSCOPY ASSESSMENT
All subjects attended for an initial screening visit during which prebronchodilator forced expiratory volume in one second (FEV1) and airway responsiveness were measured and atopic status determined. On this occasion, asthmatic subjects were asked to discontinue inhaled β2 agonists for a minimum of 6 hours. The baseline FEV1 was recorded using a dry wedge bellows spirometer (Vitalograph Ltd, Buckingham, UK). Airway responsiveness to inhaled histamine was then measured as previously described.20 Atopic status was determined by skin prick testing using the following allergen extracts:Dermatophagoides pteronyssinus, cat allergen, dog allergen, mixed grass pollens, and mixed feathers (Hollister-Steir, Elkhart, IN, USA). In addition, 10 ml peripheral blood was obtained at the time of bronchoscopy for estimation of total serum IgE (normal range <81 IU/ml).
FIBREOPTIC BRONCHOSCOPY AND BRONCHIAL BIOPSY
Bronchoscopy was performed using our previously described protocol.20 Briefly, subjects received premedication with nebulised salbutamol (2.5 mg) and ipratropium bromide (0.5 mg), intravenous atropine (0.6 mg), and intravenous midazolam. Topical lidocaine solution (10%, 4% and 1%) was used to anaesthetise the upper and lower airways. An Olympus BFIT20 fibreoptic bronchoscope (Olympus Optical Company, Tokyo, Japan) was inserted via the nasal or the oral route, and bronchial biopsy specimens were obtained from second or third generation airway carinae using FB15C biopsy forceps (Olympus Optical Company).
PROCESSING OF BRONCHIAL BIOPSY SPECIMENS
Biopsy specimens were fixed immediately by immersion in 1% paraformaldehyde solution in phosphate buffered saline (PBS; 0.01 M phosphate buffer, pH 7.4, 0.15 M NaCl) for 3–4 hours. After washing in 15% (w/v) sucrose in PBS, tissue was embedded in OCT compound mounting medium (Miles Laboratories Inc, Naperville, IL, USA) on a small cork disk and frozen by immersion in melting dichlorofluoromethane (Arcton; ICI, Cheshire, UK) cooled with liquid nitrogen. Six μm sections were cut using a cryostat, taken up onto poly-l-lysine-coated glass slides, and air dried for 60 minutes.
IMMUNOHISTOCHEMISTRY
The primary antibody to iNOS was obtained from rabbits immunised with a synthetic peptide corresponding to amino acids 54–76 of the human iNOS sequence as previously described.21Confirmation of the specificity of this antibody by Western blotting has been reported previously.21 The anti-COX-2 antiserum was raised against a 19 residue peptide (sequence NASASHSRLDDINPTVLIK) corresponding to amino acids 555–579 of the murine COX-2 sequence. The immunogen was prepared by covalent coupling of this peptide to keyhole limpet haemocyanin using glutaraldehyde. Using this antibody we were able to immunoprecipitate COX-2 from cultured human endothelial cells activated with 20 nM phorbol ester (data not shown), treatment that induced COX-2 as previously described.22 The specificity of the two antisera was further confirmed by the ability of preadsorption with the synthetic peptides (10 nmol/ml) to abolish immunostaining. The anti-iNOS and anti-COX-2 antisera were used at final dilutions of 1:1000 and 1:2000, respectively. Immunostaining was completed using an avidin biotinylated peroxidase complex (ABC) technique with nickel enhancement, as described previously.23 Sections were dehydrated, mounted in synthetic medium (Pertex, CellPath, Hemel Hempstead, UK) without counterstaining, and photographed using an Olympus BX-60 microscope.
Airway epithelial expression of immunoreactive iNOS and COX-2 was quantitated using a computer assisted image analysis system (Seescan Symphony, Cambridge, UK) as previously described.24 For each biopsy specimen the entire epithelium in three non-serial sections per antigen was assessed. The area of the epithelium, whether intact or with only the basal cells remaining, was measured after manual delineation. The area of immunostaining above an optical density threshold defined by interactive thresholding as equivalent to that of the background staining was also measured. Results were expressed as the proportion of the total epithelial area that was immunostained. Quantitation was performed by an observer (QHM) who was unaware of the clinical details of the subjects.
IN SITU HYBRIDISATION
To detect mRNA for iNOS and COX-2, sections were hybridised with32P-labelled RNA probes complementary to the appropriate mRNA. The probe for iNOS was prepared from a human colonic carcinoma cell line (DLD-1)25 by reverse transcription and the polymerase chain reaction using the primers GCG GAT CCC TCA ACA ACA AAT TCA GGT AC (forward) and GCG GAT CCA GCC GCT GGC ATT CCG CAG AA (reverse). The 200 bp cDNA fragment was cloned into the Bluescribe plasmid pBS+ and linearised sense and antisense templates were produced using Hind III and EcoRI, respectively. The COX-2 probe was prepared similarly using the primers GGC GTC AGC CAT ACA GC (sense) and ACA ACG TTC CAA AAT CCC (antisense) to yield a 228 bp cDNA corresponding to bp 128–355 of COX-2. This was cloned into the vector pCR-2 (Invitrogen, San Diego, CA, USA) and linearised sense and antisense templates were produced using Hind III and XbaI, respectively. Templates for both probes were transcribed using T7 and T3 RNA polymerases, respectively, with a mixture of 32P-labelled CTP and unlabelled ATP, GTP, and UTP.
Sections were pretreated with 0.2% Triton X-100, 0.25% acetic anhydride and water, dried, and incubated for 16 hours at 42°C with 5 ng of the labelled cRNA probe diluted in hybridisation buffer. Slides were washed in 2× standard saline citrate (SSC) with 0.1% sodium dodecyl sulphate (SDS), and in 0.1× SSC with 0.1% SDS at 42°C, and in 2× SSC at room temperature. Unhybridised single strand probe was removed with RNase (10 mg/ml) solution in 2× SSC at 37°C. After drying the slides were dipped in liquid emulsion (K5, Ilford, UK) at 45°C, dried at room temperature, and incubated in light tight cassettes for 5–10 days before development.
STATISTICAL ANALYSIS
Data for age, FEV1 % predicted, and immunoreactive iNOS and COX-2 were expressed as mean (SE). The effect of subject category was studied by one way factorial analysis of variance (ANOVA). Post hoc pairwise comparisons were made using Fisher's protected least significant difference (PLSD) test. Statistical analysis was performed using StatView 4.02 for Macintosh computers (Abacus Concepts, Berkeley, CA, USA).
Results
SUBJECT COMPARISONS
The three groups did not differ significantly in age. The FEV1 % predicted measurements did not differ significantly between the non-steroid treated asthmatic subjects and the control subjects, but were significantly lower in the steroid treated asthmatic group (p<0.01). Airway responsiveness was not measured for two of the subjects in the steroid treated asthmatic group (nos 7 and 8) as their FEV1 measurements were <40% predicted. The non-steroid treated asthmatic subjects and the remaining steroid treated asthmatic subjects had varying degrees of airway hyperresponsiveness, whereas the control subjects all had histamine PC20 measurements outside the asthmatic range (>32 mg/ml).
IMMUNOREACTIVE iNOS
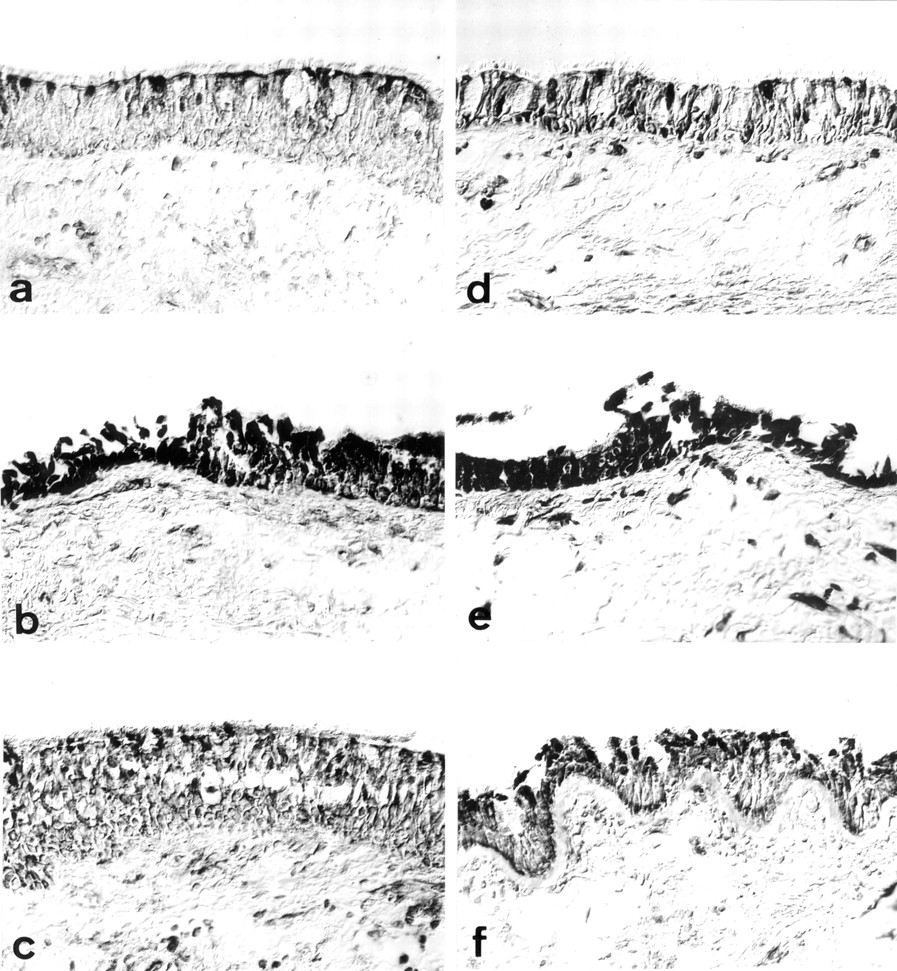
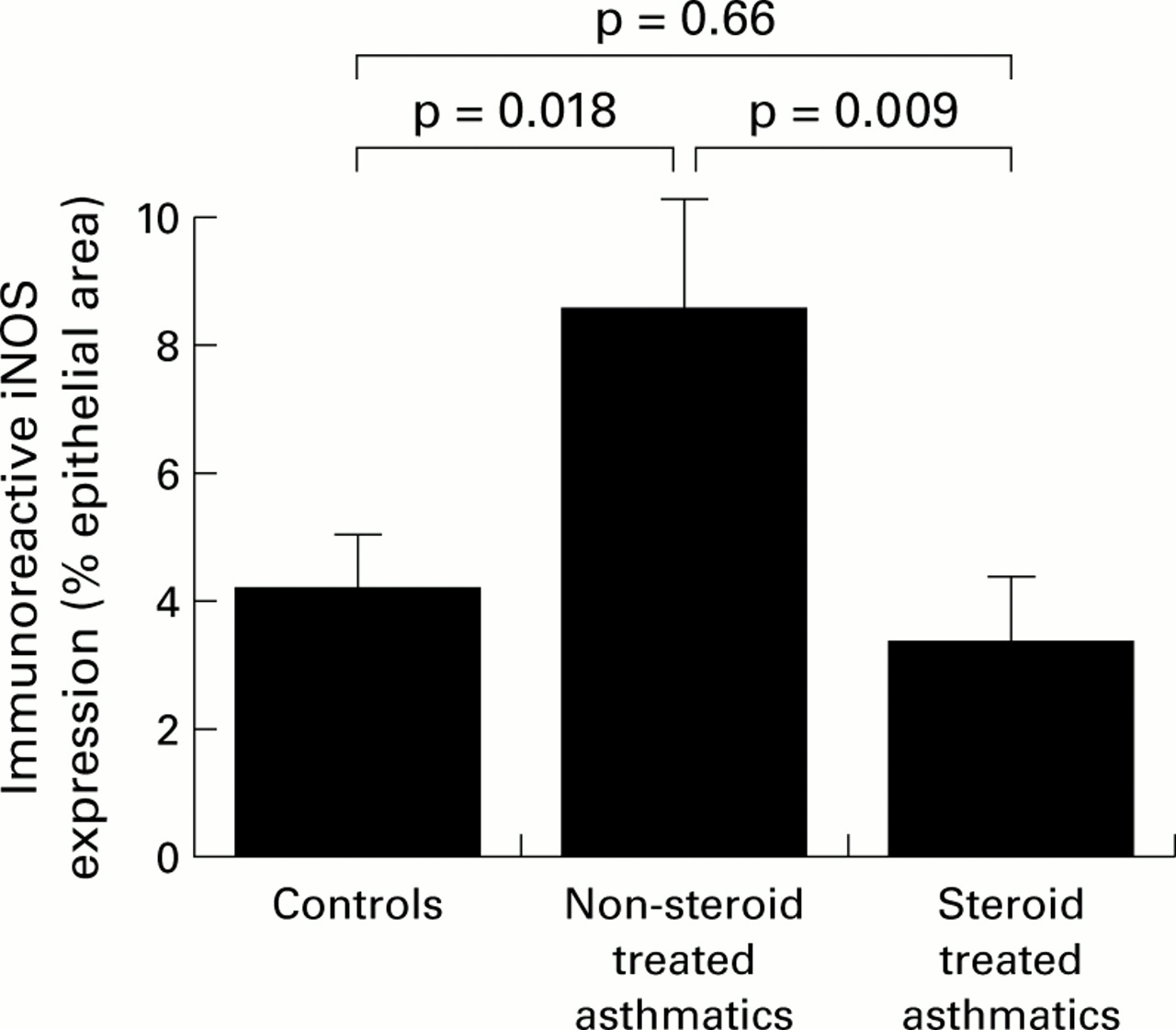
Immunostaining for iNOS was principally localised in the airway epithelium in all three subject groups (fig 1a–c). Epithelial staining appeared to be strongest in areas where focal stripping had occurred. In some instances immunoreactivity was also evident in relation to airway smooth muscle and inflammatory cells. The degree of epithelial staining was reflected in the apparent number of inflammatory cells and their intensity of staining. The proportion of epithelial area immunostained was significantly greater in the non-steroid treated asthmatic subjects (8.6 (1.8)%) than in either the non-asthmatic control subjects (4.2 (0.9)%) or the steroid treated asthmatics (3.4 (1.0)%; p=0.019, ANOVA; fig 2). There were no significant correlations between epithelial iNOS scores and FEV1 % predicted in any of the three groups. In the non-steroid treated group, but not the steroid treated group, a direct correlation with PC20 was evident (r=0.82, p=0.04).

Representative immunohistochemical staining patterns. Immunostaining of iNOS in sections from (a) a non-asthmatic control subject, (b) a non-steroid treated asthmatic subject, and (c) a steroid treated asthmatic subject, and immunostaining of COX-2 in sections from (d) a non-asthmatic control subject, (e) a non-steroid treated asthmatic subject, and (f) a steroid treated asthmatic subject.

Area of iNOS immunostaining in bronchial epithelium as a percentage of the total epithelial area. Data are expressed as mean (SE). Statistical analysis was performed by one way ANOVA (p=0.019) with post hoc testing using Fisher's PLSD test.
IMMUNOREACTIVE COX-2
The overall distribution of COX-2 immunoreactivity was similar to that of iNOS, although staining was present in a greater percentage of the epithelial area (fig 1d–f). Quantitative analysis showed that the proportion of epithelial area immunostained was significantly higher in the non-steroid treated asthmatic subjects (26.3 (4.6)%) than in either the steroid treated asthmatics (13.0 (0.6)%) or the non-asthmatic control subjects (11.6 (0.6)%; p=0.0007, ANOVA; fig 3). There were no significant correlations between immunoreactive COX-2 expression and physiological parameters in any of the three groups. Furthermore, iNOS and COX-2 scores did not correlate significantly with one another.

Area of COX-2 immunostaining in bronchial epithelium as a percentage of the total epithelial area. Data are expressed as mean (SE). Statistical analysis was performed by one way ANOVA (p=0.0007) with post hoc testing using Fisher's PLSD test.
IN SITU HYBRIDISATION
In situ hybridisation localised positive signals for both iNOS and COX-2 mRNA to the airway epithelium of non-steroid treated asthmatic subjects. Steroid treated asthmatics and non-asthmatic control subjects had no clear signal of a level above that of the background (fig4a–d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
In situ hybridisation for mRNA encoding iNOS (a, b) and COX-2 (c, d). Labelling of the epithelial cells for both probes is seen in the autoradiograms of biopsy tissue from non-steroid treated asthmatic subjects (a, c) but not steroid treated asthmatics (b, d).
Discussion
The present findings in relation to iNOS extend previous reports of expression of this enzyme in human airways. Using resected lung specimens, Kobzik et al 8 reported that the bronchial epithelium was the principal site of immunoreactive iNOS expression in large airways, in agreement with the present results. In a previous study using a semiquantitative grading system, we reported strong epithelial expression of immunoreactive iNOS in 22 of 23 bronchial biopsy specimens from asthmatic subjects, but weak or absent expression in 18 of 20 specimens from normal volunteers.26 Our present findings therefore confirm these earlier observations, and also provide the first description of increased iNOS mRNA in asthma and of the in vivo effects of corticosteroid therapy on iNOS expression.
The significance of increased expression of iNOS and NO within the airways in asthma is controversial. One interpretation would be that NO plays a protective role. For example, inhalation of NO at 80–100 ppm produces a modest bronchodilator response in asthmatic subjects with spontaneous27 or methacholine induced28bronchospasm, and endogenous NO release has been shown to reduce bronchoconstriction caused by inhaled bradykinin and methacholine in asthma.29 Alternatively, increased NO expression could contribute to the pathophysiology of the disease. In pigs, inhalation of NO induces a profound pulmonary vasodilator response30which, in asthma, could lead to oedema and plasma protein extravasation, features that contribute to airway narrowing. Furthermore, increased NO may produce cytotoxic and cytostatic effects. For example, NO is known to mediate the cytotoxicity of pertussis toxin for hamster respiratory epithelial cells,31 and has been implicated in the induction of macrophage apoptosis.32
In the present study immunoreactive COX-2 was readily detectable in bronchial epithelial cells from healthy subjects, suggesting constitutive expression of the enzyme at this site. This finding is consistent with reports of COX isoenzyme expression by cultured human airway epithelial cells33-35 and with in vivo studies using resected human lung tissue.35 ,36 Several previous studies have examined COX-2 expression in asthma, but these have produced conflicting findings. Using a similar methodology to the one described here, Sousa et al 37 reported a fourfold increase in bronchial epithelial COX-2 immunostaining in asthmatic subjects compared with non-asthmatic controls. Most of the subjects in the asthmatic group were receiving treatment with inhaled corticosteroids, generally at a relatively low dose. In contrast, Demoly et al 38 examined bronchial biopsy specimens from patients with asthma, patients with chronic bronchitis, and normal control subjects, and were unable to detect significant differences in the extent or intensity of COX-2 immunostaining between these groups. The asthmatic subjects were heterogeneous with regard to atopic status, disease severity, and smoking history, and many were receiving treatment with inhaled corticosteroids at an unspecified dose, and with other agents including long acting β2agonists. Similarly, Taha et al 39 could not demonstrate a significant increase in immunoreactive COX-2 expression in bronchial biopsy specimens from asthmatic subjects, most of whom were treated with inhaled corticosteroids, although the proportion of immunostained cells in induced sputum was increased in a separate group of non-steroid treated asthmatics. It therefore seems likely that the apparent discrepancies between these various studies relate to differences in the subject populations studied, particularly with regard to anti-asthma therapy, and perhaps also to the different methods employed for quantitation. None of the earlier studies analysed mRNA expression, and thus our present findings represent the first description of increased COX-2 expression in asthma at the mRNA level.
Both the COX isoenzymes catalyse the generation of PGH2from arachidonic acid, with subsequent metabolism potentially leading to the generation of a range of prostaglandins and thromboxanes. In vitro studies of prostanoid release by activated human pulmonary epithelial cells have indicated that PGE2 represents the major component, with PGF2α also present but TXB2 not detectable.40 ,41 Whether other enzymes in the prostanoid biosynthetic pathways are differentially regulated in asthma is undetermined. This study does, however, confirm the potential for bronchial epithelial cells to contribute to prostanoid production within the airways in vivo, and suggests that the airway epithelium may be a significant contributor to the increased levels of prostanoids in the BAL fluid of patients with asthma.
In the case of both iNOS and COX-2, the increased epithelial expression reported in the present study probably reflects exposure in vivo to proinflammatory cytokines. Expression of iNOS mRNA and protein by cultured human bronchial epithelial cells is increased by exposure to TNFα alone26 or to a combination of TNFα, IL-1, and interferon gamma (IFNγ).42 Similarly, expression of COX-2 by human pulmonary epithelial cells is increased by IL-1β and TNFα alone, and by IL-1β, TNFα, and IFNγ in combination.35 ,41 ,43 Many investigators have reported increased expression of these cytokines in asthma.1 Our biopsy findings in corticosteroid treated asthmatic subjects are also consistent with information from in vitro studies. In the case of both iNOS42 and COX-2,41 ,43 cytokine induced gene expression in cultured human pulmonary epithelial cells is inhibited by corticosteroids. These observations indicate that corticosteroid treatment in asthma is associated not just with a reduction in inflammatory cell numbers but also with suppression of inflammation-inducible gene expression. The airway epithelium is believed to represent the major target for inhaled corticosteroid therapy.
In summary, the present study indicates that airway epithelial expression of iNOS and COX-2 is increased at the level of both mRNA and immunoreactive protein in the airways of non-steroid treated asthmatics compared with non-asthmatic control subjects. In asthmatic subjects receiving regular treatment with corticosteroids, on the other hand, expression of both iNOS and COX-2 is similar to that seen in non-asthmatic subjects. These findings highlight the role of the airway epithelium both as a contributor to the inflammatory process in asthma and as a target for inhaled corticosteroid therapy in this disease.
Acknowledgments
The authors thank Professor Sir John Vane for his stimulating advice and help.