Article Text

Abstract
BACKGROUND The success of haematopoietic (bone marrow or peripheral blood) stem cell transplantation (SCT) is compromised by pulmonary complications. We hypothesised that a proinflammatory alveolar microenvironment, reflected in alveolar macrophage (AM) cytokine production, would predispose to such complications.
METHODS AM were isolated from adult SCT recipients by bronchoalveolar lavage before SCT (n=32) and during post-transplant pancytopenia (n=23). Concentrations of tumour necrosis factor (TNF)α, granulocyte-macrophage colony stimulating factor (GM-CSF), interleukin (IL)-1β, IL-6, and IL-8 in 24 hour AM culture medium were measured by enzyme linked immunosorbent assay and compared with both the occurrence of post-SCT lung disease and with subjects' previous respiratory histories.
RESULTS Eleven subjects developed lung disease within 6 months of SCT. These subjects had higher median pre-transplant AM TNFα (8 (IQR 1–8)v 2 (1–5) ng/106AM, p=0.01, median difference (D) = 3, 95% CI 0.1 to 7), GM-CSF (5 (0.7–8)v 0.2 (0.1–0.8), p=0.006, D = 4, 95% CI 0.5 to 7), and IL-6 (0.5 (0.1–1) v 0.1 (0.02–0.3), p=0.049, D = 0.3, 95% CI 0.0002 to 1) production than remaining subjects; IL-1β and IL-8 did not differ. During pancytopenia high AM GM-CSF production again predicted later lung disease (1 (0.7–9) v 0.1 (0.06–0.3), p=0.01, D = 1, 95% CI 0.1 to 6). A history of recent chest disease was associated with high AM TNFα and GM-CSF production and with post-SCT lung disease. Pre-SCT lung function was unrelated to post-SCT lung disease.
CONCLUSIONS Recent respiratory disease and persistent proinflammatory AM behaviour detectable before transplantation are associated with lung disease following SCT. These associations may prove useful in pre-transplant risk assessment.
- haematopoietic stem cell transplantation
- alveolar macrophage
- cytokines
Statistics from Altmetric.com
Marrow ablative cytotoxic therapy with autologous or allogeneic haematopoietic stem cell rescue (bone marrow or peripheral blood stem cell transplantation, SCT) is used increasingly in adults to treat haematological malignancy, solid tumours, and severe autoimmune disease,1 ,2 but complications of the procedure limit its use. Lung diseases (infective, inflammatory, or idiopathic) are among the commonest complications: 11–60% of recipients develop lung disease following transplantation. The incidence varies according to transplant type and underlying disease, but reported case mortality is 40–60%.3-5 A technique allowing pre-transplant estimation of the risk of post-transplant lung disease would therefore be clinically valuable.
In recipients of allogeneic bone marrow transplants, impaired gas transfer and increased bronchoalveolar lavage (BAL) fluid albumin levels before transplantation are associated with post-transplant pulmonary disease and mortality.6 ,7 These measures were not predictive in individual cases, but the observations suggest that alveolar abnormalities before SCT may predispose to subsequent lung disease. We hypothesised that this predisposition may be detectable as proinflammatory alveolar immune cell behaviour. Unlike other alveolar immune cells, the resident alveolar macrophages (AM) survive marrow ablative cytotoxic therapy and persist for up to 90 days after allogeneic SCT.8 ,9 We compared the proinflammatory behaviour of AM obtained from SCT recipients before transplantation and during post-transplant pancytopenia with the patients' clinical progress.
AM activity was defined using a panel of well characterised proinflammatory cytokines produced by AM and involved in infectious and inflammatory lung diseases: tumour necrosis factor α (TNFα), granulocyte-macrophage colony stimulating factor (GM-CSF), and interleukins (IL)-1β, IL-6, and IL-8.10-12
Methods
STUDY DESIGN
This prospective study was approved by Lothian health ethics committee. Adult patients attending Edinburgh's Western General Hospital Bone Marrow Transplant Unit for SCT were eligible; patients were excluded if the responsible clinicians judged them unfit for bronchoscopy and BAL. With informed written consent, subjects underwent fibreoptic bronchoscopy and BAL before starting the cytotoxic transplant conditioning regimen (B1) and again at the nadir of post-transplant pancytopenia (B2). Subjects were observed for new lung disease for 6 months after SCT. Proinflammatory cytokine production by AM from each specimen of BAL fluid was assayed by a researcher blind to the subjects' clinical progress and compared with the occurrence of post-transplant lung disease. Data from subjects with clinically apparent lung disease at the time of BAL were excluded from the analysis. The recent respiratory history and pulmonary function of each subject were compared with AM cytokine production and with post-transplant lung disease.
CLINICAL ASSESSMENT
The history, examination, chest radiograph, and pulmonary function were recorded before transplantation. The following aspects of respiratory history were regarded as relevant: current tobacco smoking, respiratory symptoms in the month before B1, chest disease requiring continuing treatment such as asthma or with residual abnormalities of chest radiography or lung function, documented parenchymal lung disease such as infection since the diagnosis of malignant disease. Pulmonary function tests included forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) by wedge bellows spirometry (Vitalograph, Buckinghamshire, UK), total lung capacity (TLC) by steady state helium dilution (P K Morgan, Kent, UK), and single breath carbon monoxide transfer factor and coefficient (Tlco and Kco, P K Morgan).
Post-transplant lung disease was defined by new radiographic opacification, or sustained tachypnoea (>25 breaths/min) with a fall in oxygen saturation (Sao 2) of >5% from baseline on two consecutive readings, occurring within 6 months of transplantation. Changes due to proven recurrent malignancy were excluded. Lung disease was sought by daily assessment while subjects were in hospital, by questioning at clinic attendances, and by medical record review at the end of the study.
BRONCHOALVEOLAR LAVAGE
Fibreoptic bronchoscopy was performed under sedation and local anaesthesia. Thrombocytopenic patients (platelets <40 × 109/l) received platelets before bronchoscopy. The middle lobe or lingula was lavaged with up to 240 ml normal saline at 37°C, with gentle aspiration after each 30 ml aliquot. Details of the methods and complications of bronchoscopy and BAL in this study are reported elsewhere.13
CYTOKINE ASSAYS
Lavage fluid was filtered through gauze. Cells were pelleted at 1000 rpm, resuspended in medium 199 (Gibco BRL, Paisley, UK), and counted under direct light microscopy. AM were isolated by adhesion to culture plates over 1 hour at 37°C. Floating cells were counted and discarded; adherent AM were re-incubated in RPMI-1640 medium (Gibco BRL) with 5% fetal calf serum (FCS) for 22 hours at 37°C. The AM number was defined as the initial cell count minus the floating cells. After incubation the medium was spun to remove cell debris (1000 rpm) then stored at –80°C. Cytokine levels in this medium were assayed in a single batch using in house sandwich enzyme linked immunosorbent assays (ELISA).
ELISA used the following incubations with 100 μl reagent/well on microtitre plates (Immulon 1B, Dynex, Chantilly, Virginia, USA). Plates were washed three times after each step with 0.05% Tween 20 in phosphate buffered saline: (1) 3 hours at 37°C with monoclonal mouse anti-human cytokine antibody (Genzyme, Cambridge, Massachusetts, USA or anti-TNFα, Immunogenetics, Belgium) in ELISA coating solution (KPL, Gaithersburg, Maryland, USA); (2) 1 hour at 37°C with blocking buffer (BSA diluent/blocking solution, KPL); (3) 18 hours at 4°C with duplicate aliquots of AM supernatant diluted both 5-fold and 20-fold in RPMI/FCS. Each plate also included log dilutions between 7.8 and 1000 pg/ml of standard cytokine solutions (Genzyme) for calibration, plus blocking buffer and blank negative controls; (4) 3 hours at 18°C with polyclonal rabbit (goat for IL-8) anti-human cytokine antibody (Genzyme) in blocking buffer; (5) 3 hours at 18°C with alkaline phosphatase conjugated donkey anti-rabbit or anti-goat IgG antibody (Jackson ImmunoResearch, West Grove, Pennsylvania, USA) in blocking buffer; (6) 1 hour at 18°C with 1 mg/ml phosphatase substrate (Sigma 104, Sigma-Aldrich, Poole, UK) in 10% diethanol amine buffer.
The results were read at 405 nm on an automated spectrophotometric ELISA reader. Undetectable cytokine levels were ascribed the value of the lower detection limit of the assay (10 pg/ml). Cytokine concentrations were adjusted according to the number of adherent AM/ml culture supernatant.
DATA ANALYSIS
Normally distributed results are quoted as mean (SD) values and were compared with the two tailed Student'st test. Other results are quoted as median (interquartile range) values and were compared by the Mann-Whitney rank sum test. Categories were compared by the χ2 test. Statistical calculations were performed with Sigma-Stat for windows 1.0 (Jandel Corporation, Erkrath, Germany). Relative risks and median differences with confidence intervals were calculated by recommended methods.14 ,15
Results
SUBJECTS
Of 60 eligible patients, five were unfit for BAL and 21 withheld consent, giving a study population of 34 (table 1). One B1 was unsuccessful and one subject was unavailable for B1, so 32 B1 procedures performed a median of 9 (range 1–32) days before transplantation are analysed. Six subjects withheld consent for B2 and four had lung disease, so 24 procedures performed a median of 6 (3–11) days after transplantation are analysed.
Characteristics of subjects undergoing bronchoalveolar lavage procedures B1 and B2
As transplant conditioning, 32 subjects received agents which have been implicated in lung toxicity: total body irradiation (10–12 Gy, n=4), melphalan (120–200 mg/m2, n=16), cyclophosphamide (4–8 g/m2, n=15) carmustine (BCNU, 300 mg/m2, n=8), and busulphan (600 mg/m2, n=4).16-19 Ten different conditioning regimens were used; because of this variety, their effects on post-transplant lung disease have not been analysed.
LUNG DISEASE
Eleven subjects (median age 44 years, 5 women; 10 B1 subjects and five B2 subjects) developed lung disease within 6 months of SCT with two fatalities (table 2). The underlying diagnoses and transplant types of these subjects were similar to the total study population: breast cancer (n=2), non-Hodgkin's lymphoma (n=2), Hodgkin's disease (n=1), myeloma (n=2), chronic myeloid leukaemia (n=1), acute leukaemia (n=2), aplastic anaemia (n=1); autologous peripheral blood SCT (n=7), autologous bone marrow SCT (n=2), allogeneic SCT (n=2).
Subjects with post-transplant lung disease and/or relevant respiratory histories: clinical details and cytokine production status
ALVEOLAR MACROPHAGE ACTIVITY
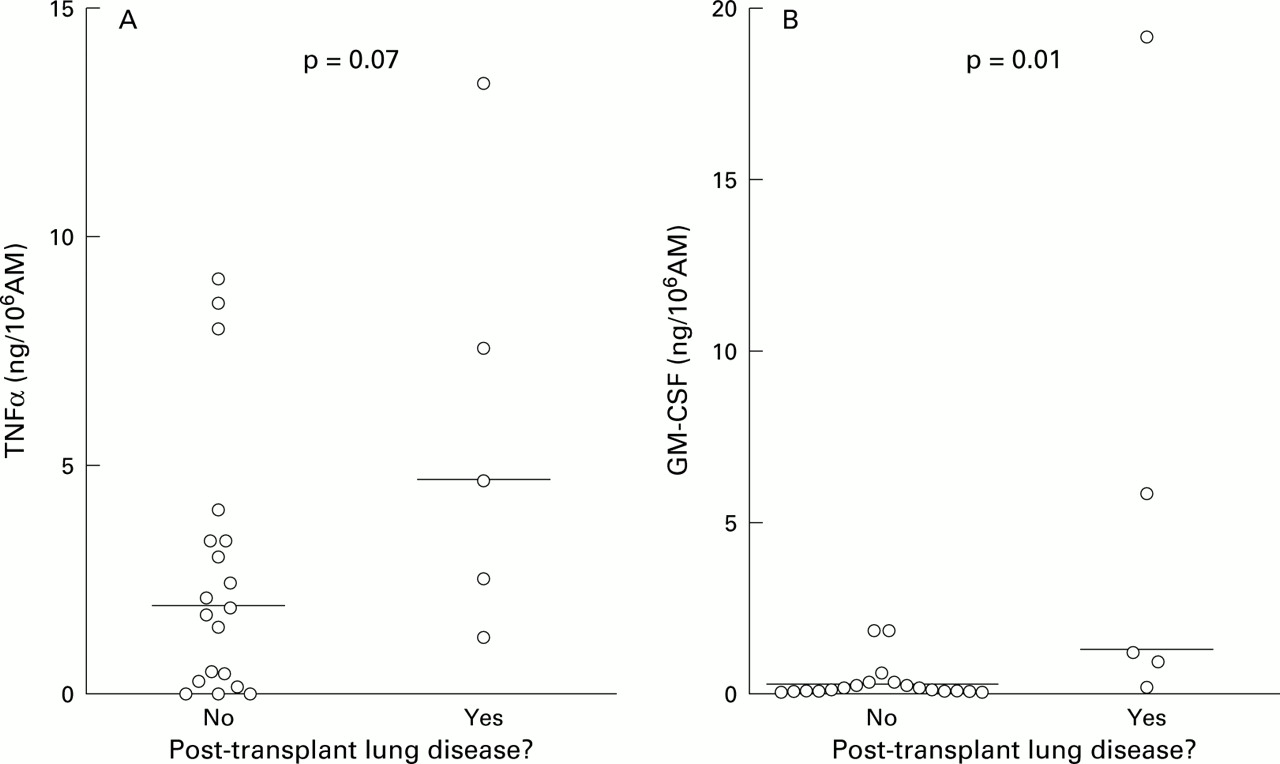
AM obtained before transplantation (B1) from subjects who developed post-transplant lung disease showed higher median TNFα, GM-CSF, and IL-6 production than those from subjects who did not develop disease (table 3). IL-1β showed a trend in this direction but IL-8 production did not differ between the groups. High B1 TNFα and GM-CSF production were strong predictors of lung disease in a categorical analysis using cut off criteria chosen retrospectively. All six subjects with pre-transplant TNFα production >7 ng/106 AM developed post-SCT lung disease (p<0.0001, relative risk (RR) 6.5, 95% confidence interval (CI) 2.6 to 16, fig1A), as did seven of nine subjects with pre-transplant GM-CSF production >2 ng/106 AM (p=0.0004, RR 6.0, 95% CI 2.0 to 18, fig 1B).
Cytokine production by alveolar macrophages (AM), divided retrospectively according to subjects' development of lung disease after stem cell transplantation (SCT)

(A) TNFα and (B) GM-CSF production by pre-transplant alveolar macrophages (AM): comparison between subjects who developed post-transplant lung disease and those who did not. Horizontal lines = median values; Δ = subjects with a relevant respiratory history; ○ = other subjects.
AM obtained during post-SCT pancytopenia (B2) displayed similar profiles, although the data were weakened by the smaller number of subjects at this time point. Subjects who later developed lung disease showed higher B2 AM GM-CSF production; there was a similar but non-significant trend in TNFα production (table 3). Production of IL-1β, IL-6, and IL-8 at B2 did not differ between the groups. Applying the criteria used in the pre-transplant analysis, two of five subjects with B2 TNFα production >7 ng/106 AM later developed lung disease (NS, fig 2A), as did both subjects with B2 GM-CSF production >2 ng/106 AM (p=0.004, RR 7.3, 95% CI 2.6 to 21, fig 2B).
{kind=link}
{kind=link}
(A) TNFα and (B) GM-CSF production by alveolar macrophages (AM) obtained during post-transplant pancytopenia: comparison between subjects who subsequently developed lung disease and those who did not. Horizontal lines = median values.
RESPIRATORY HISTORY AND PULMONARY FUNCTION
Sixteen subjects gave a relevant respiratory history (table 2). This was a risk factor for post-SCT lung disease: nine (56%) developed respiratory disease compared with two of 18 remaining subjects (11%, p=0.005, RR 5.1, 95% CI 1.3 to 20). However, B1 AM from subjects with a relevant history did not show higher median cytokine production than the remainder (respiratory history (n=15): median (interquartile range) TNFα 1.19 (1.0–8.2) ng/106 AM, GM-CSF 0.68 (0.09–8.1), IL-1β 0.45 (0.09–1.2), IL-6 0.19 (0.04–0.49), IL-8 393 (148–507); no respiratory history (n=17): TNFα 3.18 (1.6–6.1), GM-CSF 0.56 (0.07–1.2), IL-1β 0.19 (0.09–1.2), IL-6 0.15 (0.03–0.33), IL-8 502 (256–622), all p>0.2; fig1).
Since both pre-SCT AM cytokine production and respiratory history were related to post-transplant lung disease, we were concerned that these data could conceal a subpopulation of high cytokine producers with a relevant history. Categorical analysis showed that the levels of TNFα and GM-CSF production associated with a high risk of post-SCT lung disease were also associated with a relevant respiratory history. Of 15 B1 subjects with a relevant history, five had TNFα production of >7 ng/106 AM compared with one of 17 with no history of respiratory disease (p=0.047, RR 5.7, 95% CI 0.7 to 43). Seven B1 subjects with a relevant history had GM-CSF production >2 ng/106 AM compared with two with no history (p=0.03, RR 7.3, 95% CI 2.6 to 21). We could not identify any subsections of the broadly defined “respiratory history” (such as cigarette smoking) that were particularly associated with high TNFα or GM-CSF production (table 2).
Pulmonary function tests performed a median of 32 days (range 7–88) before transplantation showed no differences between subjects who developed post-transplant lung disease and those who did not (post-SCT lung disease (n=11): mean (SD) FEV1 92 (19)% predicted, FEV1/FVC 79 (10)%, TLC 91 (17)% predicted, Tlco 81 (13)% predicted, Kco 89 (14)% predicted; no lung disease (n=22): FEV1 102 (20)%, FEV1/FVC 82 (9)%, TLC 99 (14)%, Tlco 81 (19)%, Kco 87 (19)%; all p>0.2.)
Discussion
Proinflammatory AM activity detectable before transplantation and positive findings in the respiratory history were both associated with lung disease following SCT. These associations have not previously been described and, if confirmed in larger studies, could identify high risk patients before transplantation with greater confidence than other reported prognostic markers.6 ,7 ,20 These are not independent associations: many subjects who had increased AM activity also had a relevant history. In these cases the pathology reflected in the history could have caused the increased AM activity, or persistent proinflammatory AM behaviour could have predisposed to both pre-transplant and post-transplant lung disease.
An association between high pre-transplant proinflammatory AM cytokine production and subsequent lung disease is biologically plausible. The cytokines showing the association (TNFα, GM-CSF and IL-6, plus IL-1β which showed a trend in that direction) have complex interrelated functions in the initiation and maintenance of inflammation. GM-CSF, in particular, maintains a proinflammatory alveolar environment21-23 which could predispose to overt clinical disease in response to toxic or infectious challenge. In contrast, we found no relationship between IL-8 production and later lung disease. This chemokine has a specific role as a neutrophil chemoattractant in response to injury, so our subjects studied before any insult might not be expected to show differing levels.24 ,25
AM survive with little change in numbers into the post-transplant pancytopenic phase,8 ,9 so the pre-transplant AM population has continuing influence after SCT. Our data from post-SCT pancytopenic subjects were weakened by the exclusion of subjects who had already developed lung disease at the time of post-transplant BAL: this was done to avoid the confounding effects of new active disease. Despite this, we found that high AM GM-CSF production during pancytopenia was associated with later lung disease, and there was a trend in this direction for TNFα. This supports the hypothesis that there is an AM activation pattern associated with lung disease, and that this pattern persists after SCT.
We have not identified a reason for the increased AM activation shown by some of our subjects. The relationship between high cytokine production and a history of pulmonary infection or inflammation did not apply to all high cytokine producers, and most of those with a history did not show high production. A further mechanism is needed to account for persistent proinflammatory AM activity in a minority of subjects: continuing subclinical or unreported respiratory disease and genetic variability are both possibilities.
Identification before transplantation of individuals at risk of post-transplant lung disease would be clinically valuable, allowing the use of alternative treatments or of conditioning regimens with less pulmonary toxicity, and prompting heightened surveillance for post-SCT lung disease. Pulmonary function testing is widely used in this context, and large studies have shown associations between impaired function and post-transplant lung disease.6 ,17 ,26 Our impression has been that mild impairment of lung function is insufficiently specific for clinical use in predicting post-SCT lung disease, and our negative findings concerning lung function support that view. This study suggests that a positive respiratory history may be a more significant marker of risk and that, among those with a history, persistent AM activation identifies a subgroup at very high risk.
This work should be viewed as preliminary; we cannot yet recommend that all candidates for SCT should undergo assay of AM cytokine production. Our population was too small to allow comparisons with mortality, and was heterogeneous in underlying disease, transplant type, and conditioning regimen. In this respect it was typical of reports from many transplant centres, but the benefits of an invasive technique should be proven in individual patient groups to avoid unnecessary procedures. Further work is therefore needed, but this approach has the potential to reduce the exposure of patients to the pulmonary side effects of stem cell transplantation.
Acknowledgments
This work was funded by a Chest Heart and Stroke Scotland Research Fellowship awarded to Dr Whittle. The authors would like to thank Drs AC Parker, RCF Leonard, MJ Mackie, and PRE Johnson for assistance and for permission to study their patients; all the staff of the Haematology and Medical Oncology departments of the Western General Hospital; and the study subjects.