Article Text

Abstract
BACKGROUND Idiopathic pulmonary fibrosis (IPF) is characterised by subpleural fibrosis that progresses to involve all areas of the lung. The expression of transforming growth factor-β1 (TGF-β1), a potent regulator of connective tissue synthesis, is increased in lung sections of patients with IPF. TGF-β1 is generally released in a biologically latent form (L-TGF-β1). Before being biologically active, TGF-β must be converted to its active form and interact with both TGF-β receptors type I and II (TβR-I and TβR-II). TGF-β latency binding protein 1 (LTBP-1), which facilitates the release and activation of L-TGF-β1, is also important in the biology of TGF-β1.
METHODS Open lung biopsy samples from patients with IPF and normal controls were examined to localise TβR-I, TβR-II, and LTBP-1. Alveolar macrophages (AM) and bronchoalveolar lavage (BAL) fluid were examined using the CCL-64 bioassay to determine if TGF-β is present in its active form in the lungs of patients with IPF.
RESULTS Immunoreactive L-TGF-β1 was present in all lung cells of patients with IPF except for fibroblasts in the subepithelial regions of honeycomb cysts. LTBP-1 was detected primarily in AM and epithelial cells lining honeycomb cysts in areas of advanced IPF. In normal lungs LTBP-1 immunoreactivity was observed in a few AM. AM from the upper and lower lobes of patients with IPF secreted 1.6 (0.6) fmol and 4.1 (1.9) fmol active TGF-β, respectively, while AM from the lower lobes of control patients secreted no active TGF-β (p⩽0.01 for TGF-β in the conditioned media from AM obtained from the lower lobes of IPF patientsv normal controls). The difference in percentage active TGF-β secreted by AM from the lower lobes of patients with IPF and the lower lobes of control patients was significant (p⩽0.01), but the difference between the total TGF-β secreted from these lobes was not significant. The difference in active TGF-β in conditioned media of AM from the upper and lower lobes of patients with IPF was also not statistically significant. BAL fluid from the upper and lower lobes of patients with IPF contained 0.7 (0.2) fmol and 2.9 (1.2) fmol active TGF-β, respectively (p⩽0.03). The percentage of active TGF-β in the upper and lower lobes was 17.6 (1.0)% and 78.4 (1.6)%, respectively (p⩽0.03). In contrast, BAL fluid from control patients contained small amounts of L-TGF-β. Using immunostaining, both TβR-I and TβR-II were present on all cells of normal lungs but TβR-I was markedly reduced in most cells in areas of honeycomb cysts except for interstitial myofibroblasts in lungs of patients with IPF. TGF-β1 inhibits epithelial cell proliferation and a lack of TβR-I expression by epithelial cells lining honeycomb cysts would facilitate repair of the alveoli by epithelial cell proliferation. However, the presence of both TβRs on fibroblasts is likely to result in a response to TGF-β1 for synthesis of connective tissue proteins. Our findings show that biologically active TGF-β1 is only present in the lungs of patients with IPF. In addition, the effects of TGF-β1 on cells may be further regulated by the expression of TβRs.
CONCLUSION Activation of L-TGF-β1 and the differential expression of TβRs may be important in the pathogenesis of remodelling and fibrosis in IPF.
- idiopathic pulmonary fibrosis
- transforming growth factor β
Statistics from Altmetric.com
Transforming growth factor β (TGF-β), a multifunctional cytokine, is present in three isoforms in mammals.1Although TGF-βs have numerous biological effects in vitro and are ubiquitously expressed in vivo,2 the TGF-β1 isoform is most prominently expressed in numerous diseases characterised by fibrosis.3 TGF-β1 is synthesised as a large precursor protein with a pro-region and a mature region.4Intracellular processing leads to the cleavage of the pre-protein, also known as the latency associated peptide 1 (LAP-1), by the intracellular protease furin.1-4 Upon secretion, LAP remains non-covalently associated with the mature region of TGF-β1 and is then called latent TGF-β1 (L-TGF-β1), which is not biologically active unless the LAP-1 is removed or conformationally altered to yield the active 12.5 kDa dimer of TGF-β1.1-4 In some instances L-TGF-β1 can be complexed with a larger protein called the latent transforming growth factor binding protein 1 (LTBP-1).5 LTBP-1 is synthesised by a different gene from TGF-β1 and, upon binding with L-TGF-β1, it facilitates the secretion of L-TGF-β1.6 When L-TGF-β1 is complexed with LTBP-1 it is called large latent TGF-β1.6 Once LTBP-1 is extracellular it can associate with a cell surface7 or proteins of the extracellular matrix and L-TGF-β1.8 The association of L-TGF-β1 with matrix associated LTBP-1 or a cell may serve to store preformed TGF-β1 at an extracellular site where it can be released from LTBP-18by proteases such as plasmin.4 ,7-9 Once released from LAP-1, TGF-β1 can act in an autocrine or paracrine manner.
The TGF-βs mediate their effects by interacting with a serine/threonine kinase receptor complex10 ,11 in which only the TGF-β receptor type II (TβR-II), which is autophosphorylated, can bind TGF-β.11 Upon binding with TGF-β, TβR-II recruits TGF-β receptor type I (TβR-I) and phosphorylates TβR-I which in turn initiates signal transduction mediated by downstream cytoplasmic proteins, in particular the SMADS.11
IPF is a progressive and lethal fibrotic lung disease of unknown aetiology.12-15 Although the pathogenesis is not clearly understood, it is likely to be complex and to involve the interaction of many different cytokines and factors. Based on the well recognised effects of TGF-β1 on inflammation and connective tissue synthesis, we have previously used lung sections from patients with IPF and from normal controls to determine the expression of TGF-β114 ,15 and have shown by immunohistochemistry that TGF-β1 is aberrantly expressed in alveolar macrophages (AM) in early lesions of IPF.15 TGF-β1 was not present in alveolar epithelial cells or extracellularly in these regions, but in areas of advanced fibrosis and remodelling called honeycomb cysts in lung sections from the same patients, TGF-β1 was overexpressed in both the AM and in epithelial cells lining the honeycomb cysts.14 ,15 Furthermore, large quantities of matrix associated TGF-β1 were found subepithelially where there was marked fibroconnective tissue.14 ,15 TGF-β2 and TGF-β3 were ubiquitously expressed in all pulmonary cells irrespective of the presence or absence of disease.15
In the present study we show that AM obtained from patients with IPF but not normal controls secrete large quantities of active TGF-β1. In addition, in BAL fluid from the lower lobes of patients with IPF with evidence of fibrosis (as shown by high resolution computed tomographic (HRCT) scanning13) there were increased quantities of both active and latent TGF-β. BAL fluid from the upper lobes of patients with IPF contained only L-TGF-β. LTBP-1 immunoreactivity was abundant in AM and epithelial cells lining honeycomb cysts in IPF but not in alveolar epithelial cells of early lesions in IPF patients or normal lungs. In contrast, LAP-1, which identifies the presence of biologically inactive L-TGF-β1, was expressed by all pulmonary cells except interstitial myofibroblasts. The expression of TβR-I and II was ubiquitous in normal lungs, but in IPF lungs TβR-I was decreased on most cells in areas of honeycombing except for interstitial fibroblasts which expressed both TβR-I and TβR-II. These findings, in the context of our previous results, suggest that in vivo regulation of the biological effects of TGF-β1 in IPF may be affected by the release of active TGF-β1 and the spatial distribution of LTBP-1 and TβRs.
Methods
MATERIALS
The purification and specificity of monoclonal LAP-1 antibody16 and recombinant human monoclonal anti-LTBP-1 have been described elsewhere.17 Antibodies to LAP-1, LTBP-1, polyclonal antibodies to TGF-β1–3, TGF-β1, TGF-β2, TGF-β3, porcine TGF-β1 for the standard curve and polyclonal and monoclonal rabbit IgG were purchased from R & D (Minneapolis, MN, USA). Antibodies to TGF-β receptors I (V-22) and II (L-21) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
PATIENTS
The clinical characteristics of patients whose lung sections were used for immunohistochemical studies have been described in detail elsewhere.14 ,15 Briefly, lung tissue was used from paraffin embedded lung biopsy specimens obtained between 1985 and 1992 for diagnosis of diffuse pulmonary infiltrates. At the time of open lung biopsy separate sites that, on gross examination, appeared to be normal and abnormal were resected for histological diagnosis. Patients from whom AM were obtained by BAL were recruited in 1993–9 (table 1). Before recruitment the study protocol was reviewed and approved by the human ethics committee of the University of Manitoba and all patients gave signed informed consent.
Clinical characteristics of study patients
Clinical criteria for IPF (insidious onset of shortness of breath on exertion, inspiratory crackles, bilateral basal interstitial fibrosis on a plain radiograph, HRCT scan showing basal and pleural based interstitial fibrosis and, in some instances, honeycombing) were present in five patients (nos 1–5, table 1). Clubbing was detected in three of these. Pulmonary function tests were compatible with restrictive lung disease with a reduced vital capacity, diminished gas transfer (not shown), but normal flow rates. There was no evidence of joint inflammation, redness, or pain, and autoantibodies were not present. The patients did not have clinical or radiological evidence of left ventricular failure or pneumonia and were not on corticosteroids or any other immunosuppressive medications. Five patients who were being evaluated for haemoptysis or pulmonary nodules who had no evidence of inflammation or fibrosis were selected as controls (nos 6–10, table 1). None of the control patients was receiving corticosteroids or any other immunosuppressive drugs. All had pulmonary function tests which were either within normal limits or demonstrated mild to moderate airway obstruction but not restrictive lung disease, and none had HRCT evidence of interstitial lung disease.
IMMUNOHISTOCHEMISTRY
The procedure for immunohistochemistry has been described previously.14 ,15 ,18 ,19 Briefly, sections of paraffin embedded lung were treated at 58°C, deparaffinised, rehydrated, and equilibrated in Tris buffered saline (TBS) containing 0.3% Triton X-100 (Sigma). Endogenous peroxide activity was blocked using 0.3% H2O2 (Sigma) in methanol for 30 minutes at room temperature. The sections were treated with 0.1% trypsin in 0.05 M Tris, 0.02 M CaCl2 (pH 8.0), non-specific binding was eliminated by blocking with 1.5% normal goat serum (Vector Laboratories, Burlington, CO, USA) in TBS/0.5% BSA. Sections were incubated with the primary antibody or normal rabbit serum. Lung sections were pretreated with 0.5% trypsin for 15 minutes and subsequently incubated overnight at 4°C with antibodies to TβR-I and TβR-II at a final concentration of 1 μg/ml. Normal rabbit 1gG (Lipshaw, Immuno, Pittsburgh, PA, USA) was used at 1 μg/ml as a negative control. For anti-LTBP-1 a dilution of 1:2000 of the antibody or rabbit IgG as control was used and the sections were incubated for 2 hours at 37°C.20 For anti-LAP-1 a dilution of 1:100 was used or rabbit 1gG as control and the sections were incubated overnight at 4°C. In all cases the slides were incubated with biotinylated goat anti-rabbit secondary antibody (1:2000 in TBS/0.5% BSA; Vector Laboratories) followed by avidin biotin complex (ABC; Vector Laboratories diluted in TBS/0.1% BSA) for 1 hour at room temperature. Immunoreactivity was developed using the substrate 0.5% 3, 3′-diaminolunzidine tetrahydrochloride (DABA) (Sigma) in 0.1% H2O2, 0.05 M Tris, 0.85% NaCl (pH 7.4) for 2.5–5 minutes. The sections were counterstained with Gill's haematoxylin, dehydrated, and mounted with Permont (Sigma).
BRONCHOALVEOLAR LAVAGE (BAL)
Before BAL all patients with IPF had an HRCT scan. BAL was performed using a standard approach.21 Lavage samples were obtained from upper lobes where, based on the HRCT scan, there was evidence of either no disease or the presence of ground glass changes. BAL samples were also taken from the lower lobes which, based on the HRCT scan, had changes compatible with pulmonary fibrosis and/or honeycomb lung. The first 10 ml of BAL fluid retrieved was used for measuring the TGF-β content while the entire volume of fluid retrieved was used to obtain cells for culture. After centrifugation the supernatant was separated from the pellet containing inflammatory cells and the quantity of TGF-β measured and the TGF-β isoform types present were assessed using the CCL-64 bioassay as previously described.22-26 The cell pellet was washed, suspended in α-MEM, and the AM cultured to determine TGF-β levels and isoform type as previously described.
CULTURE OF MACROPHAGES
Serum free α-MEM containing gentamicin (4 mg/ml; Roussel, Montreal, Canada), fungizone (100 μl/100 ml, Gibco, NY, USA), and 0.2 % clotted bovine calf plasma (BCP) (National Biological Laboratory Limited, Dugald, Manitoba, Canada) was added to cell pellets of previously centrifuged BAL fluid.22-25 The cells were allowed to adhere for 2 hours, washed, and the adherent cells were identified as macrophages22-25 and cultured in the above media. The conditioned media were collected in the presence of the protease inhibitors lepeptin (0.5 μg/ml), pepstatin (1 μg/ml), and aprotinin (1 μg/ml) (all from Sigma, Oakville, ON, USA) and frozen at –80°C until ready for TGF-β identification.
CCL-64 BIOASSAY FOR DETECTION AND CHARACTERISATION OF TGF-β
To determine TGF-β isoforms the CCL-64 growth inhibition assay was used as described previously.22-26 Briefly, to measure the quantity of biologically active TGF-β present, neutral conditioned medium or BAL fluid was used. Total TGF-β was measured by acidifying an aliquot of the same BAL fluid or conditioned medium to activate the latent TGF-β present in the sample. The acidified BAL fluid or conditioned medium was neutralised before adding the sample to subconfluent cells (5 × 104/cells/0.5 ml) in 24 well costar dishes (Flow Laboratories Inc; Mississagua, ON, USA) 0.2% BCP, α-MEM, 10 mM Hepes at pH 7.4, penicillin and streptomycin (both 25 μg/ml). After 22 hours cells were pulsed with 0.25 μCi 5-(125I) iodo-2′-dioxyuridine (ICN Pharmaceutical, Costa Mesa, CA, USA) for 3 hours at 37°C, lysed with 1 N NaOH, and the amount of 125I-UdR incorporated was determined by counting in a gamma counter (LKB Instruments, Gaithersberg, MD, USA). Each assay was done with a standard curve using porcine TGF-β1. Confirmation of TGF-β activity or isoform identification was performed by using neutralising antibodies anti-TGF-β1, anti-TGF-β2, or anti-TGF-β3 added before the addition of conditioned media or BAL fluid.
Results
TGF-β IN ALVEOLAR MACROPHAGE MEDIUM AND BAL FLUID
The inflammation and fibrosis observed in IPF is most prominent in the lower lobes and subpleural regions; the more central portions of the lung parenchyma and upper lobes are the last to be involved as the disease progresses so, in many cases, the upper lobes of patients with IPF can appear to be relatively spared.12-15 HRCT scanning can identify regions of IPF lungs with evidence of inflammation, fibrosis, and honeycombing as well as areas that do not appear to be involved. Based on HRCT findings, BAL samples were obtained from regions with fibrosis and inflammation and from those with no apparent changes. For TGF-β to be effective it must be in its active dimeric form.1 ,4 AM obtained from patients with no inflammation or fibrosis secreted small quantities of all isoforms of TGF-β in the inactive latent form (table 2). However, AM from patients with IPF not only secreted increased quantities of TGF-β, but up to 96.6% of it was in the active form and 75% of the TGF-β from AM derived from the lower lobes of the lung was the TGF-β1 isoform (table 2). More specifically, AM from the upper lobes of patients with IPF secreted 1.6 (0.6) fmol active TGF-β while those from the lower lobes of patients with IPF secreted 4.1 (1.9) fmol active TGF-β. No active TGF-β was secreted by AM from the lower lobes of control patients. The difference in active TGF-β secreted by AM from the lower lobes of patients with IPF and from the lower lobes of control patients was significant (p⩽0.01). TGF-β from the upper and lower lobes of IPF patients was 95.8 (1.5)% and 73.3 (18.2)% active, respectively. The difference in % active TGF-β secreted by AM from the lower lobes of patients with IPF and from the lower lobes of control patients was significant (p⩽0.01). Neither the difference in total TGF-β secreted by AM from the lower lobes of patients with IPF and from the lower lobes of control patients nor the difference in active or total TGF-β in conditioned media of AM from the upper and lower lobes of patients with IPF was significant. The isoforms present in the conditioned media of AM from the upper lobes of the lung could not be determined because there was an insufficient amount of TGF-β to determine the isoform present. In all experiments TGF-β activity was confirmed by pan-anti-TGF-β1-TGF-β3 antibody.
Transforming growth factor β (TGF-β) in conditioned media (CM) of alveolar macrophages (AM)
Using the anti-LC(1–30) which detects active TGF-β1 antibody, we have previously shown that alveolar epithelial cells of normal lungs do not demonstrate the presence of immunoreactive TGF-β1 but, using the same antibody, epithelial cells lining honeycomb cysts immunostain intensely for TGF-β1.14 ,15 It was not feasible to obtain the epithelial cells to determine whether they secrete TGF-β. We therefore reasoned that alveolar lining fluid which can be retrieved by BAL should contain TGF-β secreted by epithelial cells.
Areas with minimal or no evidence of inflammation or fibrosis and areas with fibrosis and/or honeycombing were localised to the upper and lower lobes of the lungs, respectively, by HRCT scanning.13 BAL fluid from areas with no inflammation or fibrosis in patients with IPF (upper lobes) contained TGF-β in the latent form (table 3), while BAL fluid from areas with fibrosis and/or honeycomb cysts (lower lobes) contained large quantities of active TGF-β1 (69%). Lesser quantities of TGF-β2 (19%) and TGF-β3 (12%) were also present (table 3). BAL fluid from normal controls contained only L-TGF-β1 (table 3). More specifically, BAL fluid from the upper and lower lobes of patients with IPF contained 0.7 (0.2) fmol and 2.9 (1.2) fmol active TGF-β, respectively. This difference was significant (p⩽0.03). The percentage active TGF-β in the upper and lower lobes was 17.6 (1.0)% and 78.4 (1.6)%, respectively, and this difference was also significant (p⩽0.03). The total TGF-β in BAL fluid from the upper and lower lobes of patients with IPF and from the lower lobes of control patients was not statistically different. BAL fluid from the upper lobes of patients with IPF or from the lower lobes of normal controls was sufficient only to quantitate and confirm the presence of TGF-β using a pan-anti-TGF-β1–3 antibody, but was not sufficient to determine the TGF-β isoforms.
TGF-β in bronchoalveolar lavage (BAL) fluid
IMMUNOHISTOCHEMICAL ANALYSIS OF TβR-I AND TβR-II
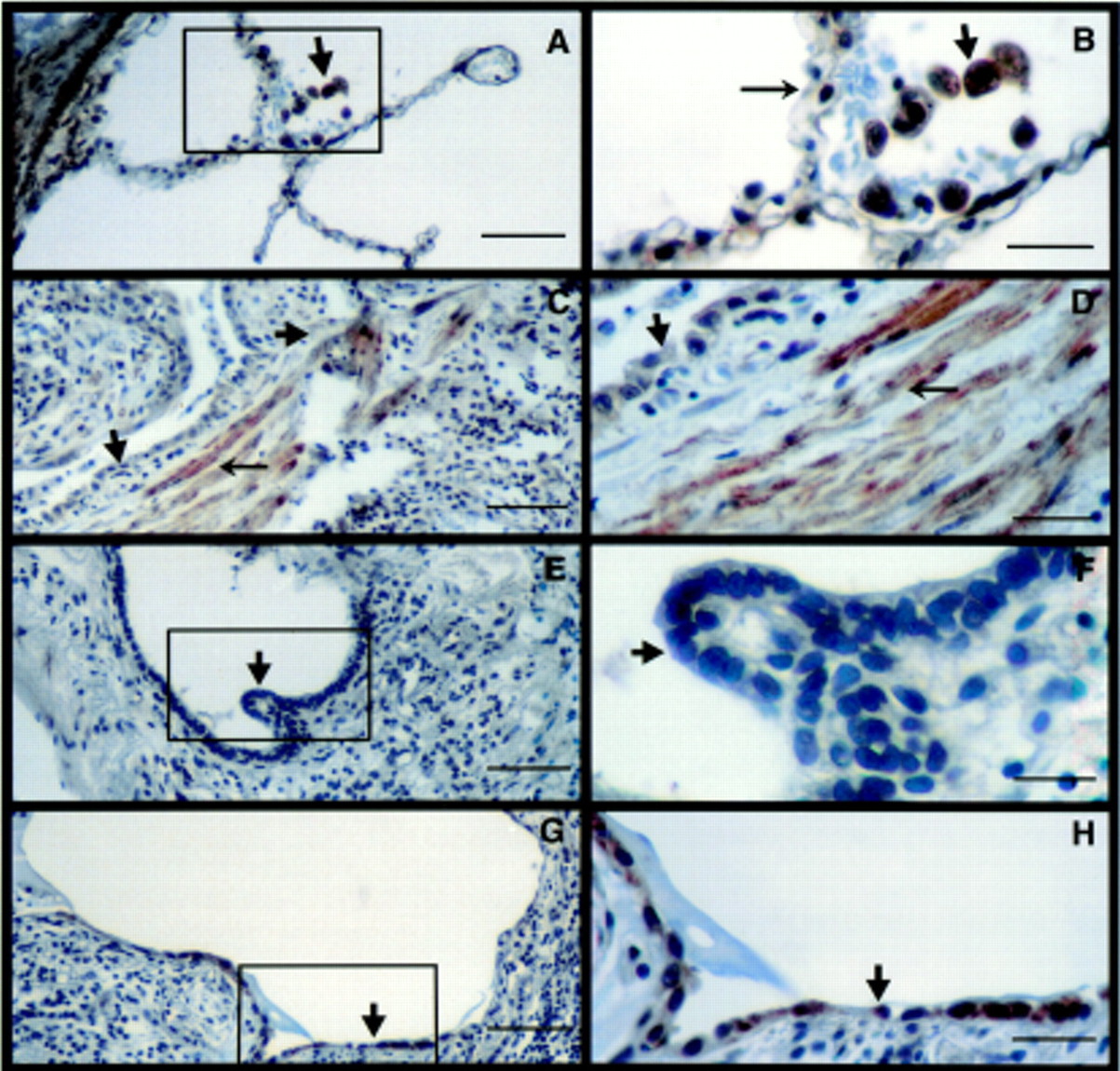
For activated TGF-β to have a biological effect, the locally responding cells require the presence of both TβR-I and TβR-II.10 ,11 We therefore determined the distribution of TβRs in the human lung sections. In normal lung the distribution of TβR-I was ubiquitous, being expressed on all lung cells including bronchial (not shown) and alveolar epithelial cells, endothelial (not shown) cells, vascular (fig 1A) and bronchial (not shown) smooth muscle cells and AM (fig 1A, B). In contrast, the lungs of patients with advanced lesions of IPF had diminished immunostaining for TβR-I throughout the entire area (fig 1C–H). In particular, the expression of TβR-I by hyperplastic type 2 alveolar epithelial cells (AECs) (fig1C, D) and other types of epithelial cells such as cuboidal cells lining honeycomb cysts (fig 1E, F) was minimal or absent. In addition, interstitial inflammatory cells including macrophages also did not express TβR-I (fig 1C–H). However, epithelial cells that appeared to be differentiated type 2 AECs expressed TβR-I (fig 1G, H). Interestingly, TβR-I was prominently expressed by spindle shaped cells likely to be fibroblasts or myofibroblasts present in the interstitium (fig 1C, D). In the normal lung TβR-II was ubiquitously present with intense staining of bronchiolar (not shown) and vascular smooth muscle cells, AECs, macrophages (fig 2A, B) and bronchiolar epithelial cells (not shown). In contrast to the paucity of TβR-I in IPF lungs, TβR-II was present in all epithelial cell types lining honeycomb cysts (fig 2C–E), AM (fig 2C, D), and fibroblasts (fig 2C, E). The immunostaining observed in figs 1 and 2 is representative of the staining in the entire biopsy section of all patients examined with IPF.14 ,15
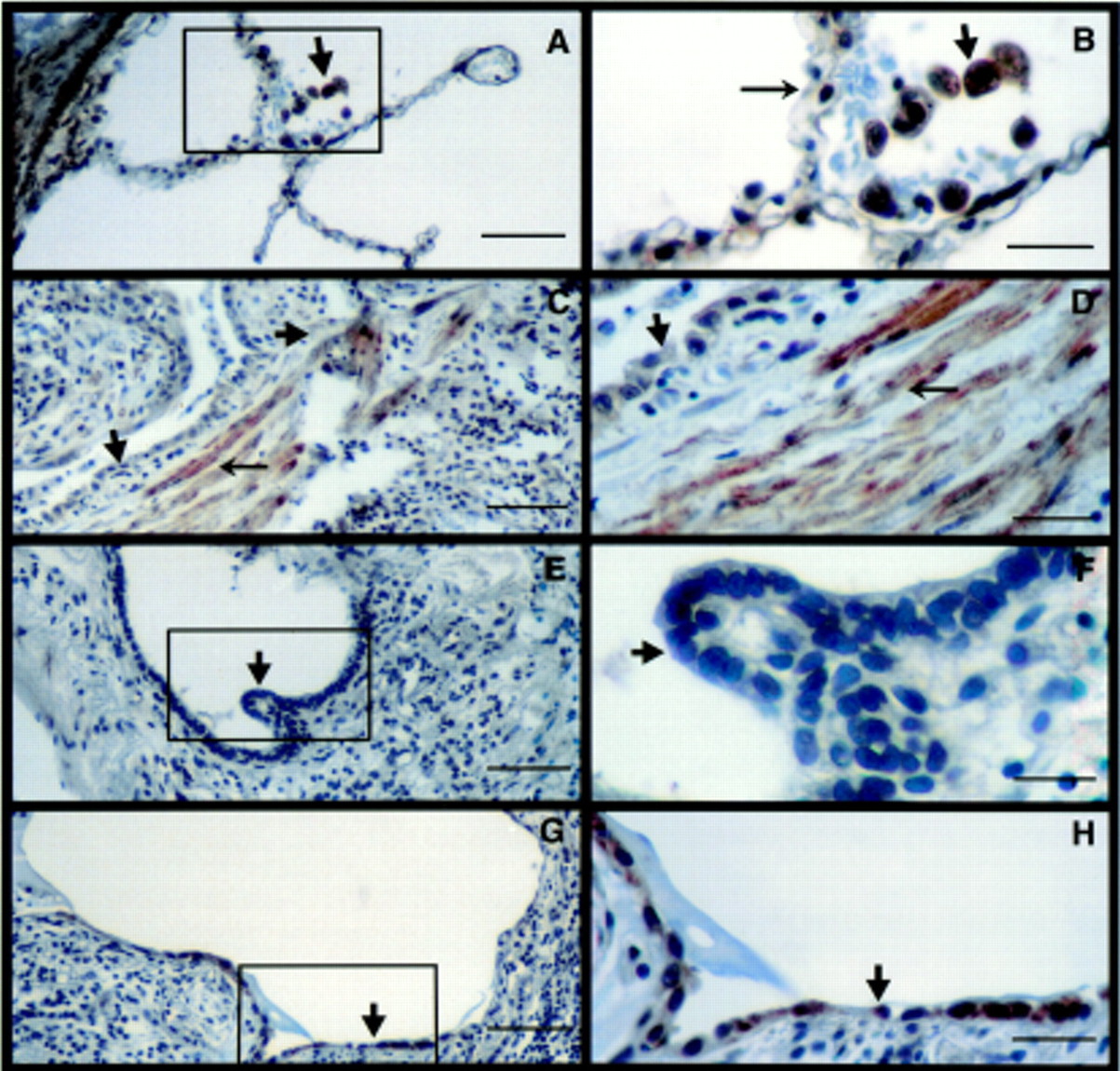
Localisation of TβR-I by immunohistochemistry. (A) TβR-I in normal lungs (bar = 50 μm). (B) Inset enlargement showing alveolar macrophages (arrowhead) and epithelial cells (arrow) expressing TβR-I (bar = 12.5 μm). (C, E, G) Distribution of TβR-I in an area of honeycomb cyst in a lung section of a patient with IPF (bar = 50 μm). There is a lack of TβR-I expression on interstitial inflammatory cells (arrowhead). (D) Inset enlargement showing lack of TβR-I on hyperplastic type 2 alveolar epithelial cells (AEC) (arrowhead) but presence of TβR-I on interstitial fibroblasts (arrow) (bar = 12.5 μm). (F) Inset enlargement confirming lack of TβR-I on cuboidal epithelial cells (arrowhead) (bar = 12.5 μm). (H) Enlargement showing presence of TβR-I on cells with type 1 AEC morphology (arrowhead) (bar = 12.5 μm). Gill's haematoxylin was used as a counterstain. The figures represent 2–3 sections from 3–4 patients.

Localisation of TβR-II by immunohistochemistry. (A) TβR-II expression in normal lung (bar = 50 μm). (B) Inset enlargement showing presence of TβR-II on alveolar macrophages (arrowhead) and AECs (arrow) (bar = 12.5 μm). (C) TβR-II expression in a lung section from a patient with IPF. Epithelial cells lining honeycomb cysts (small arrow), alveolar macrophages (arrowhead), and interstitial fibroblasts (large arrow) express TβR-II (bar = 50 μm). (D, E) Enlargements showing TβR-II on epithelial cells (small arrow), macrophages (arrowhead), and interstitial fibroblasts (large arrow) (bar = 12.5 μm). Gill's haematoxylin was used as a counterstain. The figures represent 2–3 sections from 3–4 patients.
DISTRIBUTION OF LAP BY IMMUNOHISTOCHEMISTRY
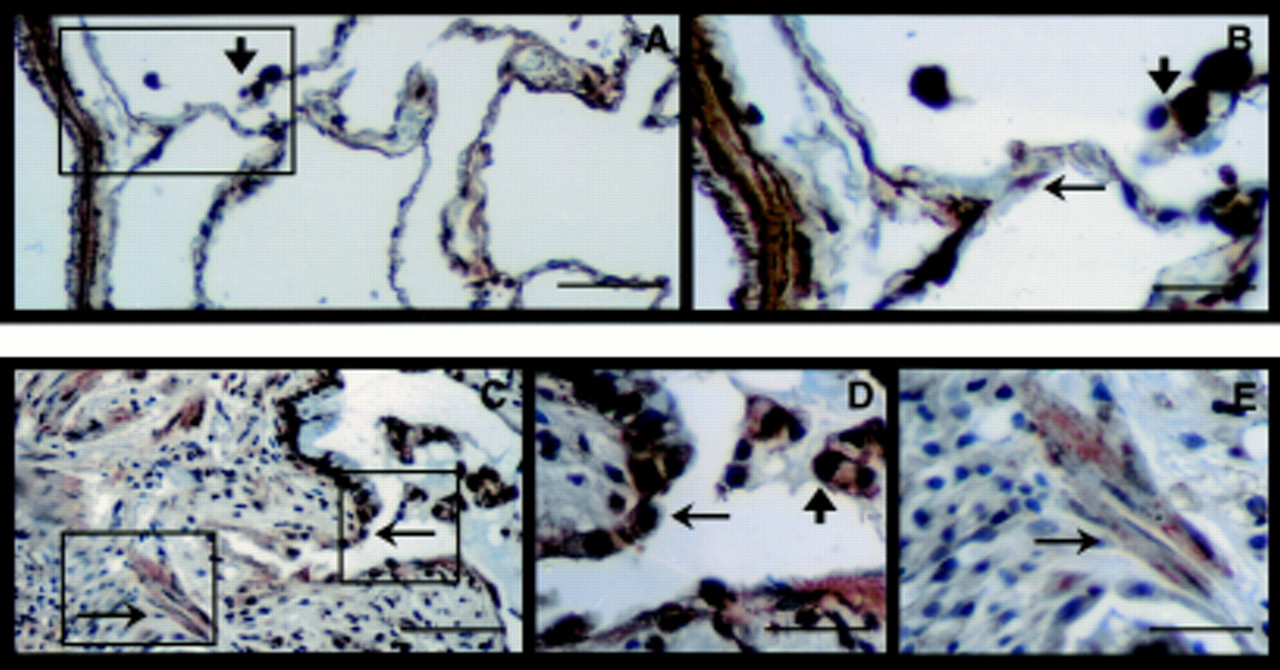
Since TGF-β is secreted in association with its LAP-1 portion, antibodies to LAP-1 should detect both the active and latent molecules.16 However, the anti-peptide antibody (anti-LC(1–30) TGF-β1 antibody) used previously identified TGF-β in an active conformation.16 LAP-1 immunostaining was observed in all cells of normal lung including bronchial and alveolar epithelial cells, bronchial and vascular smooth muscle cells and AM (not shown). Expression of LAP-1 in lung sections from patients with IPF was similar and was found in bronchial epithelial cells (not shown) epithelial cells lining honeycomb cysts, macrophages (fig 3A, B), smooth muscle cells of vessels and bronchi, and endothelial cells (not shown). In the subepithelial areas of IPF lungs where fibroblasts and extracellular matrix components were present, there was little to no immunostaining of LAP-1. The immunostaining observed in fig 3 is representative of staining found in the entire lung biopsy section of all patients examined.14 ,15

Localisation of L-TGF-β1 by immunohistochemistry. (A, B) L-TGF-β1 is present in epithelial cells (arrows) and alveolar macrophages (arrowhead) and distinctly absent in subepithelial fibroconnective tissue (bar = 50 μm for A, 12.5 μm for B). (C) Control using IgG to replace anti-LAP-1 antibody (bar = 50 μm). Haematoxylin was used as a counterstain. Each figure represents 2–3 sections from 8–9 patients.
DISTRIBUTION OF LTBP-1 BY IMMUNOHISTOCHEMISTRY
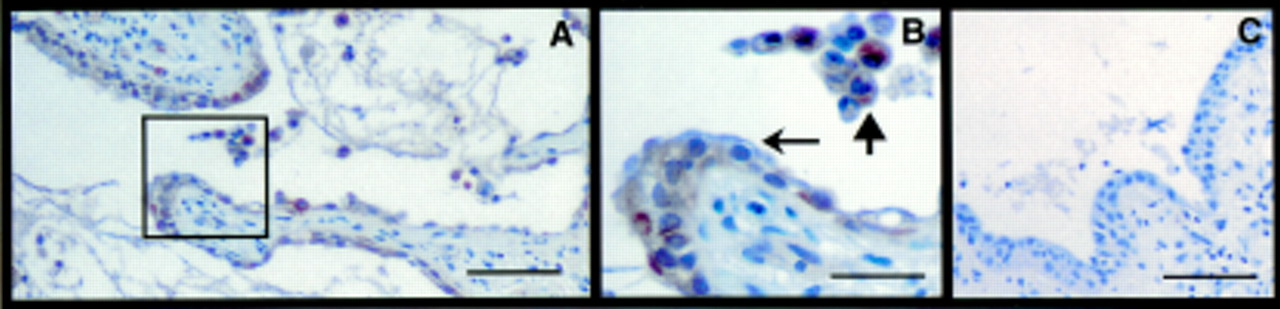
It has been shown that TGF-β can also be secreted as a large latent complex covalently bound to LTBP-1. LTBP-1 facilitates the secretion of L-TGF-β1 from the cell and the release of biologically active TGF-β1 so immunoreactivity for LTBP-1 is likely to be present in areas of TGF-β1 production and function. In normal lungs only a few AM expressed LTBP-1 (not shown). In contrast, LTBP-1 was restricted to AM and epithelial cells lining honeycomb cysts in lung sections from patients with IPF (fig 4A, B), and this immunostaining is representative of that observed in the entire lung biopsy section of all patients examined with IPF.14 ,15 Faint immunostaining of LTBP-1 was present in the smooth muscle cells of vessels and bronchi (not shown). No immunoreactivity for LTBP-1 was observed in any other cells including normal alveoli of patients with IPF (not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Localisation of LTBP-1 by immunohistochemistry. (A, B) LTBP-1 is present in epithelial cells lining honeycomb cysts (small arrow) and alveolar macrophages (arrowhead) (bar = 50 μm) and is absent in subepithelial fibroconnective tissue. (C) Control using rabbit IgG to replace anti-LTBP-1 antibody (bar = 50 μm). Haematoxylin was used as a counterstain. Each figure represents 2–3 sections from 8–9 patients.
Discussion
The pathogenesis of IPF remains largely unknown,12 ,13 but observations based on animal models of pulmonary fibrosis18 ,22-26 and lung sections from patients with IPF14 ,15 ,27 suggest that TGF-β and, particularly TGF-β1, is likely to be important in pulmonary fibrosis. Although high levels of immunoreactivity for TGF-β1, especially in AM, were previously observed in IPF, the autocrine or paracrine effects of TGF-β1 can only be manifested if the latent TGF-β1 that is secreted is converted to its biologically active form.1 ,4The findings in this study show for the first time that AM from patients with IPF secrete biologically active TGF-β1, in direct contrast with AM from normal lung which only secrete L-TGF-β. These observations are consistent with the bleomycin induced lung injury model in the rat which shows that the generation of AM derived TGF-β1 in its active form is critical to the pathogenesis of inflammation and fibrosis in this model.18 ,22-25 Our findings suggest that the release of active TGF-β1 by AM may play an important role in the inflammation and fibrosis observed in IPF.
One of the most striking features observed in the distribution of TGF-β1 in lungs from patients with IPF was the aberrant presence of TGF-β1 in epithelial cells lining honeycomb cysts.14 ,15Although we have successfully cultured rat epithelial cells in previous studies,26 we were unable to examine the release of TGF-β by explanted human alveolar epithelial cells. However, we proposed that alveolar lining fluid retrieved by BAL should contain TGF-β secreted by the epithelial cells. BAL fluid from both upper lobes where there is minimal disease and lower lobes where there is more extensive disease contained similar quantities of TGF-β. However, BAL fluid from the lower lobes contained 4–5 times more active TGF-β. The mechanism of activation of L-TGF-β1 derived from AM or present in BAL fluid in lungs from patients with IPF is not clear. However, based on animal models of fibrosis22-26and our findings using AM from patients with IPF,27 it appears that plasmin, a serine protease, mediates the release of active TGF-β1. The findings suggest that a process of L-TGF-β1 activation is present in areas of advanced lesions of IPF. It should be noted that BAL fluid could contain the secretory products of epithelial cells and also of alveolar and interstitial inflammatory cells and any serum proteins that could leak into the alveolar space. It has previously been found that no TGF-β1 was present in the interstitial inflammatory cells in lung sections from patients with IPF nor in rat lungs after bleomycin induced lung injury.14 ,15 ,18 In addition, none of the patients from whom BAL fluid was obtained had HRCT changes of alveolar or interstitial oedema to suggest that lung injury and leakage of serum proteins into the lower lobes had occurred. We have also shown that explanted rat alveolar epithelial cells constitutively secrete all isoforms of biologically active TGF-β.26 These findings suggest that AECs are a probable source of the biologically active TGF-β1 isoform that has been found to be associated with matrix in previous studies.14 ,15 It is likely that the same cells may contribute to the TGF-β present in the BAL fluid from the lower lobes of patients with IPF observed in the current study. Taken together, the presence of TGF-β isoforms in alveolar fluid may be from both AM and from epithelial cells. Based on the known effects of TGF-β on the synthesis of connective tissue,1-3 the presence of TGF-β1 in the alveolar space could result in recruitment of inflammatory cells, intra-alveolar inflammation, fibroblast recruitment, and proliferation as well as increased connective tissue synthesis by fibroblasts. All of these effects of TGF-β1 could then result in the remodelling and fibrosis that is observed in advanced lesions of IPF.12-15
In addition to the local availability of active TGF-β, the responding cell must express both signalling receptors TβR-I and TβR-II.10 ,11 TGF-β is a potent inhibitor of epithelial cell growth. In the bleomycin induced rat lung injury model we have shown that the expression of TβR-I by immunohistochemistry is markedly decreased on proliferating AECs.28 The lack of TβR-I in AECs is transient as these receptors reappear concomitantly with the cessation of proliferation of AECs 7 days after bleomycin injury.28 The downregulation of TβR-I expression by AECs after bleomycin induced injury is consistent with the resistance of the these cells to the antiproliferative effects of TGF-β demonstrated in cultures of AECs.28 In IPF honeycomb cysts are consistently characterised by the presence of type 2 AEC hyperplasia and epithelialisation by cuboidal cells of the respiratory bronchioles.12 ,14 ,15 Thus, the downregulation of TβR-I on epithelial cells lining honeycomb cysts, leading to the resistance of antiproliferative effects of TGF-β, could be integral to the repair of damaged epithelium by proliferation of epithelial cells in IPF.26 TGF-β also induces differentiation of epithelial cells.1 The reappearance of TβR-I on AECs in rats after bleomycin injury coincides with the time interval after bleomycin administration when AECs in areas of injury demonstrate the more differentiated type 1 AECs.26 ,28 Concomitantly, the expression of TGF-β3 in epithelial cells increases in type 2 AECs29 and there is increased secretion of TGF-β3.26 The presence of TβR-I on type 1 epithelial cells lining honeycomb cysts suggests that these cells would be responsive to TGF-β1 and TGF-β3. Thus, the presence of TβR-I on epithelial cells may be vital to the cessation of further proliferation and induction of differentiation which can be mediated by both TGF-β1 and TGF-β3.26 ,29 The mechanisms involved in regulating the dynamic expression of TβR-I by AECs in patients with IPF are unclear but preliminary data indicate that factors or cytokines present in rat BAL fluid after bleomycin injury such as PGE2 and IFNγ and autoregulation by TGF-β may be important in suppressing TβR-I expression on the human adenocarcinoma cell line A549 cells (D Douglas, N Khalil, unpublished observation).28 Since A549 cells are derived from human AECs, it is reasonable to propose that the decrease in TβR-I in IPF lungs may similarly respond to cytokines locally released in response to injury. Unlike AECs, the interstitial cells resembling fibroblasts express both TβRs in advanced lesions of IPF. In the rat 14 days after bleomycin injury there is increased expression of TβR-I and TβR-II on interstitial fibroblasts. Explanted fibroblasts isolated 14 days after bleomycin injury spontaneously synthesise maximal quantities of collagen.18These findings suggest that, in pulmonary fibrosis, the expression of TβRs by interstitial fibroblasts may be critical for TGF-β1 mediated connective tissue synthesis, thereby contributing to the fibrotic response in IPF. Collectively, these data suggest that the temporal expression of TβR-I by different cell types of the lungs is important in controlling the pathological changes observed in IPF.
In lungs of patients with IPF L-TGF-β1 is present in all pulmonary cells except for subepithelial and interstitial myofibroblasts. Similarly, in bleomycin induced lung injury in the rat the fibroblasts present in the lungs 4, 7, 14, and 28 days after bleomycin administration also do not express L-TGF-β1 (D Douglas, N Khalil, unpublished) or active TGF-β1.18 The lack of TGF-β1 expression by the fibroblasts may be of no consequence because matrix associated TGF-β1 is present in areas where fibroblasts were observed in IPF lungs14 ,15 and after bleomycin induced injury.18 These observations suggest that, in fibrotic lung diseases, TGF-β1 is available to interstitial fibroblasts which express both TβRs and can respond to TGF-β1 in a paracrine manner by synthesising connective tissue proteins.
It is interesting that, in areas of advanced disease in IPF, LTBP-1 is consistently expressed in AM and epithelial cells lining honeycomb cysts. However, in normal lungs from patients with IPF or from patients with no fibrotic disease, LTBP-1 was observed in some AM but not in epithelial cells. The presence of LTBP-1 and TGF-β (both mature and latent) in AM and epithelial cells suggests that these cells synthesise large latent TGF-β1 in IPF. Flaumenhaft et al showed that LTBP-1 of the LTBP-1/L-TGF-β1 complex can associate with the cell surface and may be important in increasing the efficiency with which plasmin activates L-TGF-β1.7 Since both AM18 ,23-25 and epithelial cells26generate active TGF-β1 and express LTBP-1, it is conceivable that LTBP-1 is important in IPF in the conversion of L-TGF-β1 to the active TGF-β1 seen in areas of fibrosis in IPF lungs. It has also been found that LTBP-1 associates with the extracellular matrix.8 ,9 L-TGF-β1 complexed with matrix associated LTBP-1 may be important as a reservoir of TGF-β1 that can be released by proteases such as plasmin.9 No LTBP-1 was observed in the subepithelial regions where matrix associated TGF-β1 had been previously demonstrated,14 ,15 which suggests that TGF-β1 may be associated with the extracellular matrix in IPF lungs, but by a mechanism that does not involve LTBP-1. Alternatively, it is possible that LTBP-1 complexed with L-TGF-β1 is present subepithelially but, when LTBP-1 is bound to the extracellular matrix, the epitopes exposed are not recognised by the antibodies to LTBP-1 used in this study.
It has previously been shown that TGF-β3 is important in the pathogenesis of pulmonary vascular diseases.30 In contrast, our data indicate that the TGF-β1 isoform is important in the pathogenesis of pulmonary fibrosis.25 TGF-β1 has numerous biological effects and the expression of TGF-β1 and its receptors is ubiquitous. The regulation of TGF-β function in both physiological and pathological processes depends on its ability to be activated and the expression of both signalling receptors on target cells in a temporally dynamic fashion. We conclude that activation of L-TGF-β1 and the differential expression of TβR-I may be important in the pathogenesis of remodelling and fibrosis in IPF.
Acknowledgments
This work was supported by the Medical Research Council of Canada and The Lung Association of Newfoundland & Labrador. The authors thank Dr Ganesh Raghu for reviewing the manuscript, Ms Carol Whitman for her technical assistance, Ms Vicki Brown for the immunohistochemistry, Dr Kevin Craib for the statistical analysis, and Ms Carolin Hoette for help in word processing.