Article Text

Statistics from Altmetric.com
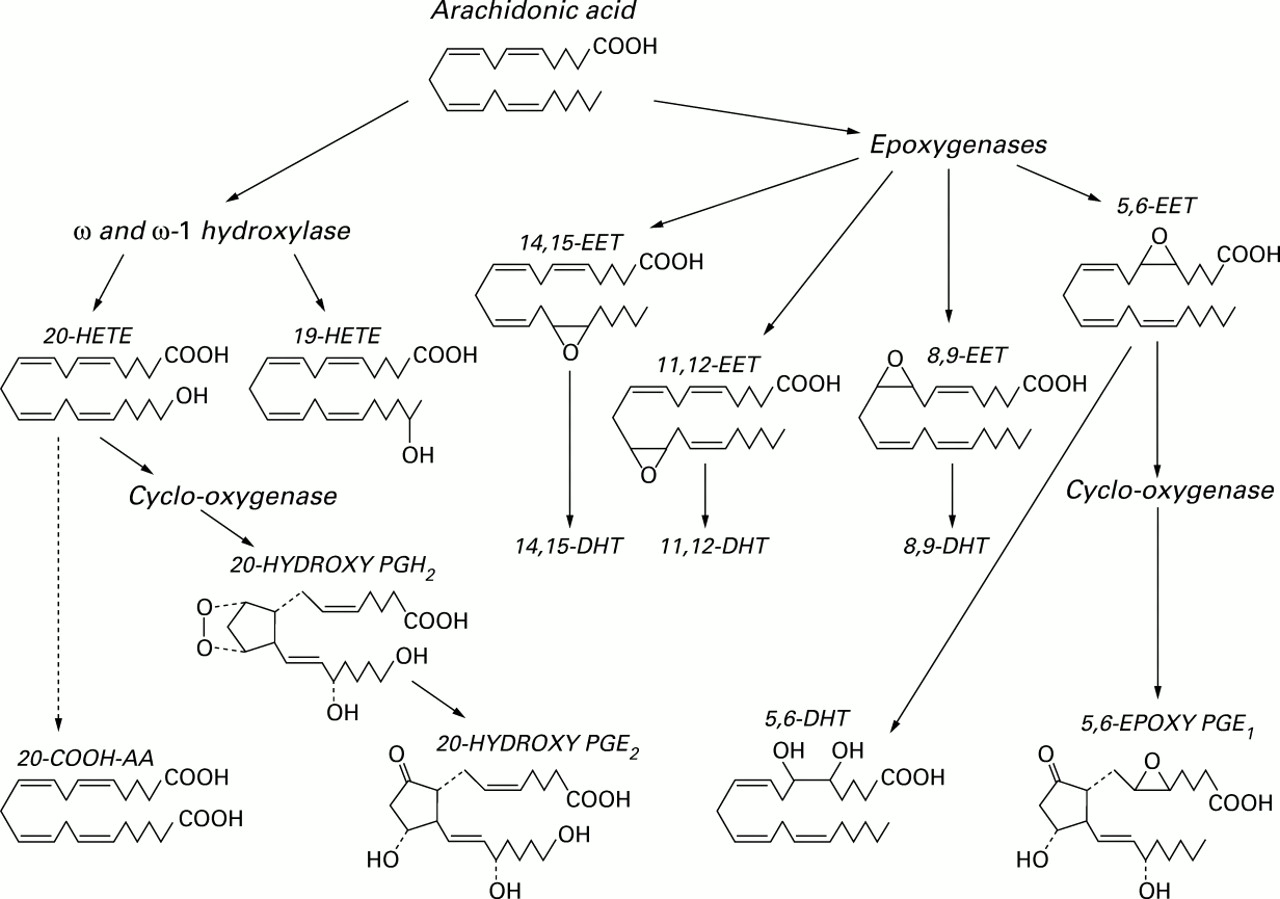
The first reports on the metabolism of arachidonic acid (AA) by cytochrome P450 (CYP) mono-oxygenases appeared in 1981.1 ,2 The biochemical studies of Capdevila and colleagues have provided the “ground substance” for all future studies. Our initial report in 1984 on the metabolism of AA via CYP pathways by the rabbit medullary thick ascending limb (mTAL) provided evidence that AA metabolism in this segment of the nephron was primarily via the CYP pathway.3 Previously the mTAL was thought to lack the biosynthetic machinery for metabolising AA because of negative immunocytochemical evidence regarding cyclo-oxygenase (COX) capabilities. A subsequent study identified two principal CYP derived AA products generated by the rabbit mTAL, one inhibiting Na+-K+-ATPase and the other relaxing blood vessels.4 Two principal CYP products—20-hydroxyeicosatetraenoic acid (20-HETE) generated by ω/ω-1 hydroxylases and 11,12-epoxyeicosatrienoic acid (11,12-EET) generated by epoxygenases—had been identified in a study of renal zonal CYP derived AA metabolites (fig 1).5 No one then could have anticipated the rise of 20-HETE to its position of pre-eminence among renal eicosanoids, one that functions as a key component in both tubular and vascular mechanisms essential to the regulation of renal haemodynamics and extracellular fluid volume. The first study that pointed to an essential role for 20-HETE in the kidney indicated that it modulated the Na+-K+-2Cl– cotransporter in the mTAL, the target for furosemide (frusemide) and the other “high ceiling” diuretics.6 Our companion study had identified 20-HETE as the principal product of AA metabolism in mTAL.7
{kind=link}
Arachidonic acid (AA) metabolism by cytochrome P450 dependent mono-oxygenases to ω- and ω-1-hydroxyeicosatetraenoic acids (HETEs), epoxyeicosatrienoic acids (epoxides, EETs), and dihydroxyeicosatrienoic acids (diols, DHTs). 20-HETE and 5,6-EET can be converted by cyclo-oxygenase to analogues of prostaglandins.
20-HETE is the pre-eminent renal eicosanoid. The protean properties of 20-HETE—vasoactivity, mitogenicity, and modulation of transport in key nephron segments—serve as the basis for its essential roles in the regulation of the renal circulation and electrolyte excretion and as a second messenger for endothelin-1 (ET-1) and mediator of selective renal effects of angiotensin II (AII). Renal autoregulation and tubular glomerular feedback (TGF) are mediated by 20-HETE through constriction of preglomerular microvessels, particularly afferent arterioles.
The synthesis by Falck and by Ortiz de Montellano of authentic standards of CYP-AA metabolites and specific inhibitors of CYP enzymes that spare COX and lipoxygenases was essential to the progression of studies,8-10 particularly as they relate to analysis of homeostatic mechanisms in terms of involvement of CYP-AA products. Selective inhibitors of ω and ω-1 hydroxylases that generate 20- and 19-HETEs, and epoxygenases generating 5,6-, 8,9-, 11,12- and 14,15-EETs are now available which allow deletion of one or other of the two major pathways of CYP dependent AA metabolism.11This capability is important in defining the functional contribution of either enzyme pathway as the products of these pathways frequently have opposing effects on the circulation.12 ,13
Features of CYP related 20-HETE metabolism
COX TRANSFORMATION OF 20-HETE
The most extensively studied in terms of transformation by COX is 20-HETE, the principal renal CYP-AA product (fig1).14 ,15 It is converted by COX to a vasoconstrictor prostaglandin H2 (PGH2) analogue (20-OH PGH2) that undergoes additional transformation by isomerases to vasodilator/diuretic metabolites (20-OH PGE2, 20-OH PGI2) and vasoconstrictor/antidiuretic metabolites (20-OH TXA2, 20-OH PGF2α).16
MODULATION OF CYP ENZYME ACTIVITY BY NO
Nitric oxide (NO) exerts a tonic inhibitory influence on CYP metabolism of AA.17 Removal of the suppressant effects of NO by inhibiting NO synthase (NOS) will result in marked perturbations of renal function resulting from increased production of 20-HETE.18 ,19 On the other hand, increased production of NO will inhibit 20-HETE synthesis.20 These interactions operate at the renal tubular and vascular levels and are important in setting the level of activities of TGF, K+ channels, and renal vasomotion.16 ,21 ,22
STORAGE OF CYP-AA PRODUCTS
HETEs and EETs can be stored in tissue lipids23 and released in response to hormonal stimuli as, for example, AII stimulated, receptor mediated hydrolysis of phospholipids (PLs).24 In contrast, prostaglandins are not stored, being synthesised on demand. Preformed HETEs and EETs bound to lipids represent a significant reservoir in those tissues in which they have been measured—namely, the liver,23 kidney,24and platelets.25
Vascular mechanisms involving 20-HETE
The localisation of ω hydroxylase to the preglomerular microvessels, the vascular segment primarily responsible for effecting changes in renal vascular resistance, and the biological properties of 20-HETE—namely, constriction of preglomerular microvessels in nanomolar concentrations26—argue for it mediating renal autoregulation. This was put to the test by demonstrating that 17-octadecynoic acid (17-ODYA), an inhibitor of CYP-AA metabolism, abolished renal autoregulation.27
The afferent glomerular arteriole, in addition to its contribution to renal autoregulation, is the effector limb of TGF that regulates the glomerular filtration rate by producing changes in preglomerular arteriolar resistance in response to solute/NaCl delivery to the TAL. 20-HETE is an essential component of TGF that may act on both limbs of the mechanism—the afferent limb related to the signal initiated by the macula densa and its transmission and the efferent limb related to constriction of the preglomerular arterioles.22 Francoet al 28 had anticipated that a non-COX, non-lipoxygenase AA metabolite was a component of TGF. They had reported that potentiation by AA of TGF was not affected by inhibition of COX or lipoxygenase, suggesting involvement of either AA directly or via a metabolite of AA generated by an undefined oxygenase. Zou et al 22 examined the relationship of 20-HETE to TGF, prompted by studies indicating that 20-HETE constricts the afferent arteriole and is a principal product of AA metabolism in both the afferent glomerular arteriole and the mTAL, the latter sharing many properties with the contiguous macula densa. Potentiation of TGF in response to AA administration into the tubular fluid perfusing the loop of Henle was blocked by inhibition of ω hydroxylase with 17-ODYA and restored in the face of ω hydroxylase blockade by the addition of 20-HETE to the tubular fluid. Thus, a major component in the mechanism underlying TGF had been identified—20-HETE mediates changes in preglomerular arteriolar resistance and, perhaps, participates in initiating the signal arising in the macula densa. As AII increased 20-HETE synthesis by preglomerular microvessels, it was assumed that AII induced gain of TGF would occur, and this was found to be the case.29
NO, another potentially important component in TGF, can be integrated into this construct as a negative modulator of TGF, in view of the localisation of NOS within both the macula densa and afferent arteriole and the functional effects of NO on TGF—namely, in response to dietary salt, NO blunted TGF.30 The negative modulatory effect of NO on TGF may be accounted for by suppression of 20-HETE synthesis—that is, production of NO by the macula densa and/or afferent arteriole can moderate constriction of the afferent arteriole by suppressing 20-HETE formation.20 As 20-HETE constricts the afferent arteriole and possibly functions in the signal transduction pathway of TGF, deletion of 20-HETE by inhibiting ω hydroxylase should inactivate TGF and this was found to be so.22 This construct is oversimplified as it excludes several factors such as adenosine and thromboxane A2(TXA2) which can modify TGF.29 ,31
Regulation of glomerular afferent arteriolar tone
The ability of preglomerular microvessels, particularly the afferent arteriole, to generate AA metabolites by the CYP pathway is directly related to the key role of these blood vessels in TGF and renal autoregulation. Further, preglomerular microvessels serve as a potential paradigm for the systemic and regional vasculatures as they are the most intensively studied blood vessels. The preglomerular microvessels also generate AA products via both COX and lipoxygenase pathways, and each pathway can be activated selectively by vasoactive hormones. The complexity of hormonal interactions with oxygenases that metabolise AA in preglomerular microvessels is evident on reviewing the effects of AII on afferent arteriolar tone. AII constricted the rat afferent arteriole by stimulating PLA2 to release AA that was transformed by lipoxygenase to vasoconstrictor AA products, 12- and 15-HETEs, the second messengers for the renal vasoconstrictor and other actions of AII.32 Inhibition of epoxygenases potentiated the constrictor response to AII, indicating that an EET—possibly 11,12-EET—counteracted AII induced constriction of the afferent arteriole. This effect of AII on epoxygenase product formation by rabbit afferent arterioles has been related to an AT2receptor mediated response.33
Increased transmural pressure in preglomerular microvessels elicited formation of 20-HETE by stimulating PLC,34 whereas stimulation by AII of PLA2 produced the lipoxygenase products 12- and 15-HETEs.32 Norepinephrine (noradrenaline) also constricted the rat afferent arteriole but independently of stimulating PLA2 and activation of lipoxygenases.32 Norepinephrine induced constriction was augmented by COX inhibition, suggesting that a vasodilator metabolite of AA, presumably PGE2 and/or PGI2, opposed the constrictor action of norepinephrine. Thus, afferent arteriolar responses to AII and norepinephrine, when analysed in terms of relationships to AA products, disclosed involvement of all three pathways of AA metabolism in modulating/mediating vascular responses to the vasoactive hormone. The complexity increases on further analysis of the effects of AII on the rabbit afferent arteriole in the presence of blockade of the AT1receptor.33 A vasodilator response to AII was uncovered that was unaffected by inhibition of either NOS or COX, but was abolished by blockade of the AT2 receptor as well as by inhibiting epoxygenases with miconazole. The EET responsible for AII induced glomerular afferent arteriolar dilatation was thought to be 11,12-EET as it was the most potent vasodilator epoxide of the afferent arteriole.35
It is important to recognise that, in the studies on the regulation of preglomerular microvessels reviewed here, the outcome and conclusions are greatly influenced by species (rat versus rabbit) and by experimental preparations that differ by including (or excluding) tubular and glomerular elements as well as by perfusing preglomerular blood vessels with either blood or artificial perfusate. For example, Arima et al 36 used rabbit afferent arterioles devoid of tubular elements and perfused with artificial medium at 60 mm Hg and found that an increase in afferent arteriolar tone provided by inclusion of norepinephrine in the perfusate was required to demonstrate constriction of afferent arterioles by 20-HETE. The importance of the attached juxtaglomerular apparatus and/or glomerulus to the actions of AII on the afferent arteriole have been emphasised by Weihprecht et al. 37 The study of Imig and Deichman,32 on the other hand, was conducted in vitro with rat juxtamedullary nephrons perfused at 100 mm Hg with reconstituted blood—that is, under conditions that simulated those considered physiological.
General statements can be made, based on the above analysis, regarding the renal vascular effects of hormones as modified by eicosanoids. Arachidonate metabolites act as second messengers and modulators of vasoactive hormones. Eicosanoid/hormonal interactions vary according to the vascular segment, experimental conditions, species, and sex. Within the vasculature, longitudinal variations in oxygenases and their products are evident as is differential localisation of CYP and COX in blood vessels viewed cross sectionally—for example, ω hydroxylase in vascular smooth muscle and epoxygenase in the endothelium. The interactions of vasoactive hormones and CYP dependent AA metabolism show a high degree of selectivity relative to a given agonist acting on a particular segment of the renal vasculature and to the eicosanoid released by the hormone that mediates or modulates the action of the hormone. Findings based on isolated structures should be retested in an integrated experimental preparation in which the renal vasculature and tubules are represented. Observations made on isolated tissues, cultured cells, and cell homogenates are limited by the absence of paracrine and endocrine influences that in situ greatly affect the response of the cell/tissue. The relationship of an individual effect of a hormone on a renal cell or tissue to the resultant multiple effects of that hormone on renal function has been summarised by Cogan for AII, but it applies to vasoactive hormones generally: “. . . the interconnected, dynamic control of circulatory and tubular transport processes is clearly important in the complex governance of renal function attributable to angiotensin II”. The operative words are “interconnected” and “governance”.38
The lung
The pulmonary CYP-AA pathway in the rabbit has been characterised by Zeldin et al.39 The predominant CYP dependent AA metabolites were EETs and their hydration products, the vicinal diols, dihydroxyeicosatrienoic acids (DHTs), 5,6-EET being generated in greatest quantities by lung microsomal fractions. Polyclonal antibodies prepared against purified CYP 2B4 epoxygenase isoform completely inhibited EET formation, indicating that CYP 2B4 is the principal constitutive AA epoxygenase in the rabbit lung. The predominant HETE was 20-HETE. EETs and DHTs were detected in rabbit bronchoalveolar lavage fluid; 14,15-EET was found in greatest quantities (40% of total), 11,12- and 8,9-EETs accounting for about 30% each. As 5,6-EET is highly labile it could not be quantified, although its stable hydration product 5,6-DHT was present in abundance (41% of total DHTs) in the bronchoalveolar lavage fluid. Rabbit lungs were considered to be “unique in their selectivity for epoxidation of AA at the 5,6 position”.
The biological activities of CYP-AA metabolites were examined on cylindrical segments of guinea pig hilar bronchi precontracted with histamine. 5,6- and 8,9-EETs each reduced, and 20-HETE increased, bronchial smooth muscle tone. It should be noted that the principal pulmonary epoxygenase isoform (CYP 2B4) has been localised to cells within the rabbit airway, a finding which occasioned the suggestion that CYP derived pulmonary EETs might be involved in the regulation of bronchial smooth muscle tone.40 Studies on vascular smooth muscle have underscored the importance of EETs as regulators of the activity of Ca2+ activated K+ channels, a site where one or more EETs antagonise the actions of 20-HETE resulting in hyperpolarisation of vascular smooth muscle and vasodilatation. Indeed, EETs, either individually or collectively, are considered to be prime candidates for endothelial derived hyperpolarising factors (EDHFs).41 An excellent case has been made by Fultonet al 42 that 5,6-EET is the EDHF mediating the vascular response to bradykinin in the rat heart.
The relevance of the similarity of EETs as agents of relaxation of vascular smooth muscle to their action on airway smooth muscle has been tested by Dumoulin et al.435,6- and 11,12-EETs were examined for their effects on bovine airway smooth muscle tone and on reconstituted Ca2+ activated K+ channels. The EETs relaxed precontracted bronchial spiral muscles in a dose-dependent manner. However, to demonstrate the relaxing effect of 5,6- and 11,12-EETs it was necessary to denude the epithelium, which suggests that the epithelial layer constituted a barrier (possibly biochemical) to the EETs. As these effects of the EETs were attenuated by iberiotoxin, an inhibitor of Ca2+activated K+ channels, the latter was considered to be the target of the EETs. Furthermore, the EETs were considered to activate airway smooth muscle by interacting directly with the Ca2+activated K+ channels. Thus, the EETs were shown to activate reconstituted Ca2+ activated K+channels independently of a G protein linkage.
In conclusion, CYP-AA metabolites have the capacity to act as regulators of smooth muscle tone and responsiveness that has important implications for understanding pulmonary function in health and disease. CYP-AA products are essential components of interactions involving several hormonal systems that have central roles in circulatory homeostasis, including angiotensins, endothelins, NO, and cytokines.44
Acknowledgments
This review was made possible by NIH grant RO1 HL25394. The author thanks Melody Steinberg for preparation of the manuscript and editorial assistance.