Article Text

Statistics from Altmetric.com
Biological effects of glucocorticoids
Inflammatory diseases such as asthma and rheumatoid arthritis are characterised at the molecular level by chronically increased expression of multiple cytokines, chemokines, kinins and their receptors, adhesion molecules, and inflammatory enzymes such as inducible nitric oxide synthase (iNOS) and the inducible cyclooxygenase (COX-2).1 At the cellular level, inflamed regions show a substantial influx of various inflammatory cells, arterial dilation, increased blood flow, plasma protein leakage, and oedema whilst, in the case of chronic asthma, substantial remodelling of the airways is observed involving excessive smooth muscle proliferation. However, these parameters of inflammation are effectively reduced by treatment with glucocorticoids by both direct and indirect mechanisms.2 ,3 For example, the reduced eosinophilia following glucocorticoid treatment in asthmatic subjects arises by direct promotion of eosinophil apoptosis and indirectly by suppressing receptor expression and production of cytokines or growth factors.4 These include factors such as interleukin (IL)-3, IL-5, granulocyte-macrophage colony stimulating factor (GM-CSF), and eotaxin which are involved in eosinophil maturation, recruitment, and survival. Similarly, glucocorticoids reduce T cell proliferation and increase T cell apoptosis via mechanisms that are at least partly the result of inhibition of the T cell growth factor, IL-2.5-8 Likewise, monocyte apoptosis is increased and influx of other infiltrating inflammatory cells is also repressed.2 ,9 Again, this is partly caused by reduced expression of adhesion molecules, both on migrating and target cells, as well as reduced expression of cytokines and chemokines from sites of inflammation.
Therapeutically, the ability to suppress a number of inflammatory indices makes glucocorticoids among the most potent anti-inflammatory agents currently available for the treatment of chronic inflammatory diseases such as asthma.2 ,3 The clinical efficacy of synthetic glucocorticoids such as prednisolone or dexamethasone stems from their ability to mimic natural glucocorticosteroids. Bodily insults, including inflammation, pain, infection or even mental stress, lead to activation of the hypothalamic-pituitary-adrenal (HPA) axis. These stimuli cause excitation of the hypothalamus, which responds by releasing corticotropin releasing hormone (CRH) (also known as corticotropin releasing factor, CRF). CRH then acts on the anterior pituitary to induce synthesis and release of adrenocorticotropic hormone (ACTH). ACTH in turn stimulates the adrenal cortex to release glucocorticoids such as cortisol. Once within the blood, cortisol is transported to target organs where it elicits numerous metabolic effects including increased blood glucose levels, stimulation of gluconeogenesis in the liver, and the mobilisation of both amino and fatty acids (fig 1). However, in addition to these metabolic effects, glucocorticoids are also potent endogenous immunological suppressors. Thus, whilst the anti-inflammatory power of synthetic glucocorticoids derives from endogenous anti-inflammatory mechanisms, the clinical usefulness of these drugs is limited by HPA insufficiency and effects on bone metabolism in addition to the metabolic effects listed above. In this respect, it is often stated that the metabolic effects of glucocorticoids result from increased transcription of genes such as tyrosine aminotransferase (TAT) and phoshoenolpyruvate carboxykinase (PEPCK),10-13whereas the anti-inflammatory properties are attributed to negative transcriptional effects on inflammatory gene expression.12-14 However, this may not wholly be the case.
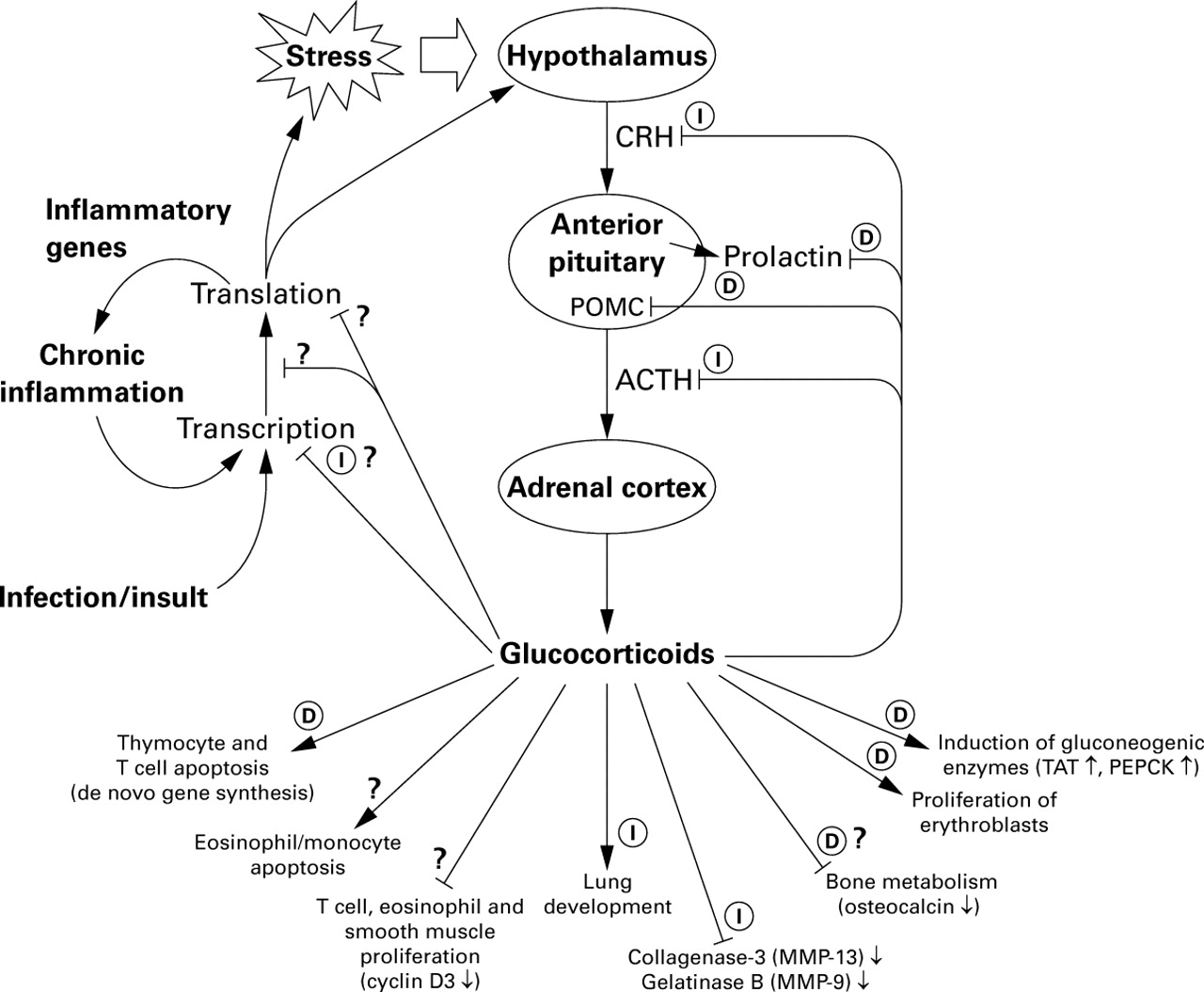
Effects of glucocorticoids on the hypothalamic-pituitary-adrenal (HPA) axis. This scheme shows the sites of synthesis and action of the main HPA hormones and the targets of glucocorticoid action (see text for details). Based on analysis of dimerisation defective mice many of the effects of glucocorticoids are labelled as either dependent on (D) or independent of (I) GR DNA binding. Question marks indicate uncertainty as to the mechanism of action. Abbreviations are to be found in the text. Adapted from Reichardt and Schutz.13
Classical mechanisms of glucocorticoid action
It is generally believed that most, if not all, the effects of glucocorticoids on cells are mediated via the glucocorticoid receptor (GR). This 777 amino acid protein was cloned in humans in 1985 and is a member of the superfamily of ligand regulated nuclear receptors.15 In common with other family members, GR has a modular structure whose principal functions—transactivation, DNA binding, and ligand binding—are localised to specific domains (fig2A).16-19 In addition, alternative mRNA splicing results in a second GR isoform, GRβ, that is defective in steroid binding and can act as a dominant negative inhibitor of GRα.15 ,20 ,21 However, this inhibition seems to require increased GRβ expression relative to GRα and a clear functional role has not yet been confirmed.

Structure of the glucocorticoid receptor. (A) Linear representation of the 777 amino acid glucocorticoid receptor showing the principal domains. DBD = DNA binding domain; LBD = ligand binding domain; τ1 and τ2 = the two activation domains; NT = amino terminal; CT = carboxy terminal. (B) Enlargement of part of the DBD showing the amino acid sequence (single letter codes) of the two zinc fingers and the dimerisation loop (in bold). Numbering of both the human and rat receptors is given. The A to T mutation at position 458 that gives rise to the dimerisation defective receptor is shown.
In the absence of ligand, GR is predominantly maintained in the cytoplasm as an inactive multi-protein complex. This consists of two hsp90 molecules plus a number of other proteins including the immunophilins p59 and calreticulin.19 Entry of glucocorticoids into the cell and subsequent binding to the ligand binding domain (LBD) of GR leads to a conformational change in the receptor. This causes dissociation of the multi-protein complex and allows nuclear translocation of GR by virtue of the nuclear localisation sequence within the DNA binding domain (DBD). Once within the nucleus, GR binds DNA sequences known as glucocorticoid response elements (GREs) to activate transcription of responsive genes (referred to as transactivation) (table 1, fig 3A).10 ,19 ,22Consistent with the palindromic nature of this motif, GR binds classical GRE sites cooperatively as a homodimer. This requires interactions between a group of five amino acids, known as the dimerisation or D loop, which is located within the DNA binding domain of each GR molecule and is essential for dimerisation and transcriptional activation (fig2B).28
GRE and nGRE sites in glucocorticoid responsive genes
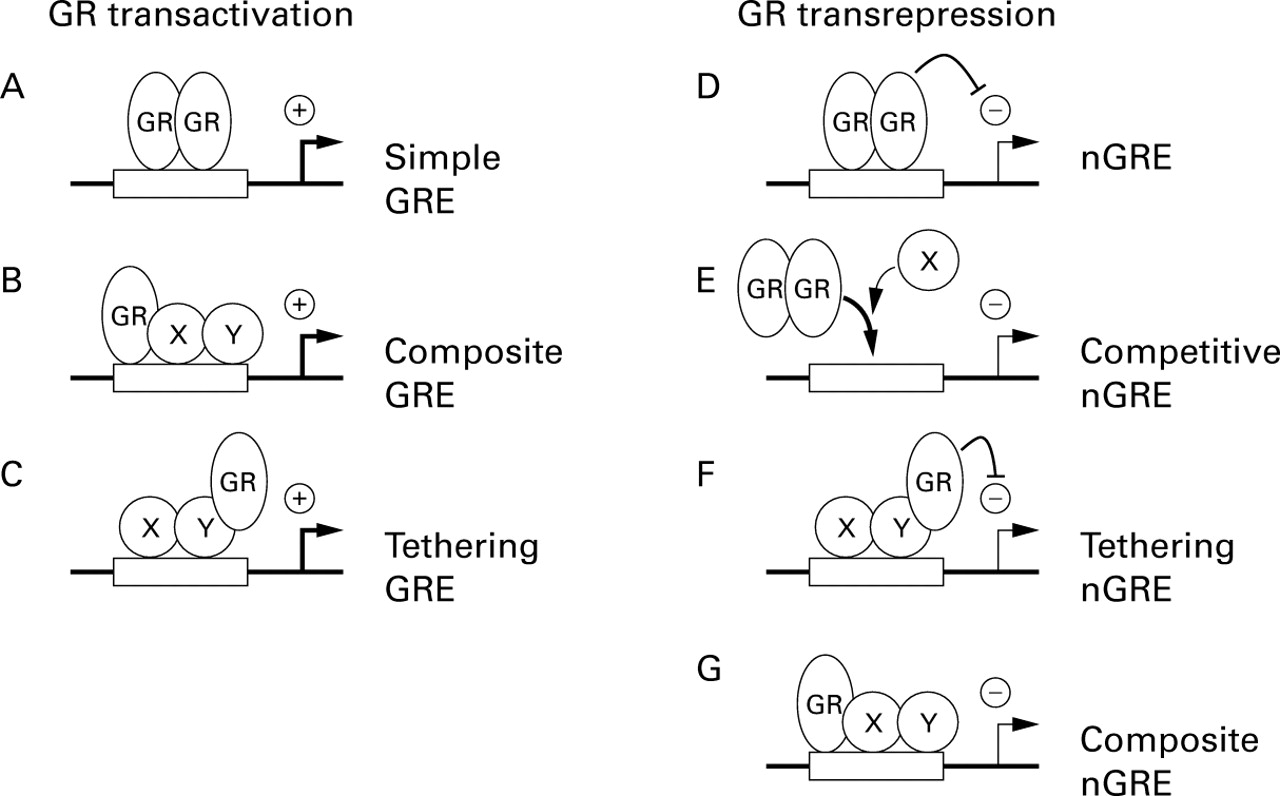
Models of glucocorticoid receptor transcriptional modulation. (A) Homodimers of GR bind cooperatively to classical GRE sites to activate transcription. (B) Interaction of GR with a second transcription factor can activate transcription from composite binding sites in a manner that involves DNA binding of both factors. (C) Interaction of GR with a second transcription factor may result in activation of transcription in a manner that does not require DNA binding of GR. (D) Homodimers of GR repress transcription from a simple nGRE. (E) At a competitive nGRE, binding of GR to the GRE site prevents binding of factors that are required for transcriptional activation and therefore causes transcriptional repression. (F) Interaction of GR with a second transcription factor may result in repression of transcription in a manner that does not require DNA binding by GR. (G) Interaction of GR with a second transcription factor can repress transcription from composite binding sites in a manner that involves DNA binding of both factors.
Genes that are known to be upregulated by glucocorticoids and play a role in resolution of inflammation include lipocortin I and p11/calpactin binding protein which are both involved in suppressing release of arachidonic acid.29 ,30 In addition, β2-adrenoreceptors,31 ,32 secretory leucocyte protease inhibitor (SLPI), and the decoy IL-1 type II receptor are also upregulated by glucocorticoids.33 ,34However, the induction kinetics for these proteins is generally slow—for example, over a 24–48 hour period—which suggests a role in the longer term anti-inflammatory effects of glucocorticoids. Thus, positive GR-dependent transcriptional mechanisms are not generally considered to explain the more rapid (0–12 hours) repressive effects of glucocorticoids on inflammatory genes.12 ,14 ,35 ,36
Negative GREs and DNA binding dependent transrepression
Following on from the characterisation of positive GRE sequences was the postulation that the negative regulation of transcription (referred to as transrepression) by glucocorticoids occurred via negative GRE sites (nGRE).22 In practice, the existence of the nGRE has remained controversial as the consensus binding site is variable and they are only described for relatively few genes.13 ,22 One major physiological function of glucocorticoids is negative feedback inhibition of the HPA axis via repression of CRH and ACTH expression (fig 1). Indeed, one promoter to be described as repressed by glucocorticoids was that for the ACTH precursor gene, pro-opiomelanocortin (POMC).37 In this case, multimers of GR were shown to bind a nGRE and to cause repression of transcription.38 Whilst the exact mechanism for this repression was not characterised, it was speculated to involve either protein-protein interactions with other factors on the POMC promoter or direct inhibition due to steric hindrance as a result of the close proximity to the TATA box and transcription start site (fig 3D and E). However, in addition to conferring glucocorticoid dependent repression, this region is also necessary for basal POMC transcription and also overlaps with a site that is involved in promoter activation.39 ,40 Thus, binding of GR to the nGRE may block binding of positive factors and thereby cause transcriptional repression (fig 3E).40 In addition, subsequent studies have attributed the dexamethasone dependent repression of the POMC promoter to tethering mechanisms akin to that for AP-1 (below), as well as to repression of factors that are induced by CRH and mediate the CRH induction of POMC.41 Whilst initially these studies appear to be conflicting, it is important to remember that the conditions used and the method of analysis have a significant bearing on the outcome. It is therefore likely that negative regulation of POMC expression is, in fact, achieved by multiple transrepressive mechanisms (see below).
Another gene to be described as glucocorticoid repressible was that for the glycoprotein hormone α subunit gene.42 This promoter is positively regulated by the cAMP response element binding protein (CREB) and contains overlapping binding sites for both CREB and GR. Thus, DNA binding by GR is proposed to inhibit transcriptional activation directly by preventing binding of CREB (fig 3E). However, as this site shows only modest homology to a consensus GRE, it is possible that GR binding is weak and the repressive mechanism more truly resembles that for AP-1 (below).12 A further example of repression by virtue of overlapping sites for GR and other transcription factors is the osteocalcin promoter.27 ,43In this case a nGRE site was described that overlapped the TATA box (table 1). Thus, binding of GR may prevent subsequent binding of the basal transcription factor, TATA binding protein (TBP). As TBP plays an important role in recruitment of RNA polymerase II to the TATA box,44 this effect will result in transcriptional repression. Similarly, a region in the bovine prolactin gene that binds GR and confers nGRE activity also acts as constitutive positive enhancer.45 Thus GR dependent repression could involve competition between factors that bind this site or neutralisation of positive activation functions by the additional binding of GR to the nGRE (fig 3D and E). In each of these cases DNA binding by GR is required for repression (fig 1).
Transrepression without DNA binding
Many inflammatory genes that are repressed by glucocorticoids are transcriptionally regulated by factors such as nuclear factor-κB (NF-κB) and AP-1.3 ,14 As nGRE sites are not generally found in the promoters of inflammatory genes, alternative mechanisms are proposed to account for the glucocorticoid dependent repression of these genes. One such mechanism is thought to arise from interaction between GR and transcriptional activators. As this effect does not require direct binding of GR to DNA, the term “tethering GRE” is often used to describe these elements (fig 3F). This phenomenon was first described for AP-1 and was thought to involve direct protein-protein interactions between GR and AP-1.46-48Functionally, the consequence of these interactions is mutual repression of both AP-1 and GR dependent transcription.46-48 Initially, reduced binding to the respective DNA recognition sites of each factor, as a result of interaction with the other, was thought to mediate this effect.46 ,48 However, no change in AP-1 DNA binding in nuclear extracts or in vivo by DNA foot printing was observed in glucocorticoid treated cells, indicating that GR mediated repression occurs via a direct effect on transcriptional activation.47 ,49
Like AP-1, glucocorticoids are able to repress the transcriptional activation by NF-κB via a direct interaction of GR with NF-κB.50 ,51 Again, this interaction was initially thought to prevent NF-κB from binding to its cognate recognition sites and thereby cause repression of transcription.51 ,52In addition, an alternative mechanism of repression of NF-κB by dexamethasone was proposed.53 ,54 This involved upregulation of the cytoplasmic NF-κB inhibitor, IκBα, to prevent nuclear translocation and DNA binding of NF-κB by retention of NF-κB heterodimers in the cytoplasm. However, a number of problems exist with this explanation. In most cell types induction of NF-κB by stimuli such as IL-1β or tumour necrosis factor (TNF)-α results in a total loss of IκBα within 5–10 minutes.55 Thus, even highly elevated levels of IκBα protein would only be expected to prevent NF-κB activation temporarily. Furthermore, as the IκBα gene is itself regulated by NF-κB, reduced NF-κB activity may be expected to reduce IκBα promoter activity and, indeed, this effect has recently been reported.56 ,57 Finally, at times that are most relevant to inhibition of inflammatory gene transcription—that is, shortly (<2 hours) after stimulation—dexamethasone has little or no effect on induction of NF-κB DNA binding by TNF-α, lipopolysaccharide (LPS), or IL-1β in endothelial or epithelial cells.55 ,57-60 In addition, the above studies failed to show any substantial increases in IκBα expression as a result of glucocorticoid treatment. Notwithstanding this, experimental conditions involving longer incubations in the presence of glucocorticoid—for example, after six hours of co-treatment with the stimulus or extended pretreatment with glucocorticoid—may cause repression of NF-κB DNA binding.55 ,61 However, these observations, whilst confirming the existence of such repressive mechanisms, have little bearing on the immediate repression of pro-inflammatory genes and probably relate to a longer term dampening effect of glucocorticoids on inflammatory processes. This conclusion is supported by Hecket al 62 who showed that any glucocorticoid dependent increase in IκBα synthesis could be dissociated from repression of gene transcription and was neither required nor sufficient for downregulation of NF-κB transcriptional activity. Consequently, the role of GR in the repression of NF-κB DNA binding and induction of IκBα may be variable and is probably dependent on the cell type and stimulus. It therefore appears that repression of NF-κB dependent transcription, like that for AP-1, primarily occurs subsequent to or downstream of DNA binding and involves GR interfering with the transcriptional activation process itself.63
Multiple transrepressive mechanisms may act in concert
In considering the various repressive mechanisms involving standard, competitive, tethering, or other nGRE sites, it is worth noting that these schemes could be viewed as variations or continuations of essentially similar mechanisms (fig 3D–G). In a given experimental context, the investigator will generally only detect the predominant mechanism involved. Thus, for example, in the case of competition for binding sites (fig 3E), GR directly binds DNA and prevents binding of factors that are necessary for transcriptional activation. However, this binding will undoubtedly involve steric (or other) hindrance of additional (unknown) factors, which may also contribute to the repression in a manner depicted in fig 3D. In addition, binding of GR to a competitive nGRE (or any other site) is also to some extent stabilised by interactions with surrounding factors that are themselves in contact with the DNA. The repressive interaction will therefore also show characteristics of a tethering or composite type nGRE (fig 3F and G). Likewise, the situation where GR binds strongly to DNA (fig 3D or E) and is stabilised by weak interactions can be contrasted with a situation where GR shows very weak or no interaction with DNA and is tethered in position by interactions with other DNA binding factors (fig 3F). Furthermore, it is possible to view the composite nGRE as an intermediate between these two extremes. Thus, at nGRE sites, GR prevents the positive transcriptional response via mechanisms that are likely to involve multiple protein-protein interactions that prevent activation of the basal transcription complex by activating transcription factors or co-factors. In any given case it is likely that a combination of these mechanisms will contribute to the overall (trans-) repressive effect (fig 3D–G).
Mutuality of transcriptional repression
One feature that is common to GR mediated repression of both AP-1- and NF-κB dependent transcription is the mutuality of the effect. Not only is GR capable of repressing AP-1 and NF-κB dependent transcription, but AP-1 and NF-κB may also repress GR dependent transcription.46-48 ,50 ,51 ,64 One explanation for this effect is that GR competes with AP-1 or NF-κB for components of the transcriptional apparatus that are limiting and necessary for gene activation. In this respect the transcriptional co-activator molecule, CREB binding protein (CBP), which plays a part in the activation of transcription by numerous transcription factors/transcriptional activators, has attracted much attention.65 ,66 This large protein is capable of binding to and co-activating with many activators including CREB, AP-1, STATs, and NF-κB, as well as steroid hormone receptors such as GR, progesterone receptors, thyroid hormone receptors, and the retinoid receptors.65 ,67-71 The function of CBP and similar proteins is currently under investigation but is thought to involve linking transcriptional activators to the basal transcription complex.71 In addition, CBP possesses an intrinsic histone acetytransferase activity.72 ,73Thus, recruitment to the promoter of proteins such as CBP and p300/CBP associated factor (P/CAF), which binds to CBP and also shows histone acetyltransferase activity,74 ,75 will cause acetylation of histone proteins and result in derepression of the chromatin structure.73 This facilitates transcription by allowing access to and unwinding of the DNA by components of the transcriptional machinery and represents a further regulatory control point.71 ,76 In this respect, members of the nuclear hormone receptor family that are involved in transcriptional repression have been shown to exist in a repressor complex along with histone deacetylase activities.77 If this observation also holds true for GR, then recruitment of histone deacetylases to the promoters of inflammatory genes may result in loss of acetyl groups from the core histone proteins. This would lead to transcriptional silencing and would provide a novel mechanism whereby glucocorticoids can repress transcription.
The concept of disassociating steroids
Leading on from the belief that the predominant anti-inflammatory effects of glucocorticoids derive from inhibition of transcription (transrepression), whereas the metabolic effects derive from positive transcriptional effects (transactivation), a number of investigators have sought to separate these functions using various mutant GR constructs and specialised “dissociating” steroids. Deletion studies with mutant GR constructs have shown that the steroid binding domain, the activation domains, and a functional DNA binding domain are necessary for efficient hormone inducible transcription from GRE containing promoters.17 However, the ability to repress an AP-1-dependent promoter (tethering type transrepression) was localised to the DNA binding domain (DBD) of GR.64 Indeed, a number of point mutations in the DBD were identified that maintained the ability to transactivate, but had lost the ability to transrepress, AP-1 mediated transcription.64 Furthermore, these two functions were clearly distinguished by the D loop mutant, A458T (fig2B).64 This mutation results in a protein that is defective in cooperative dimerisation and is unable to bind classical GRE sites or activate GR dependent transcription from GRE containing promoters.28 ,64 As the ability to transrepress AP-1 dependent transcription was unaffected, these data support the hypothesis that direct binding of GR to DNA is not required for transrepression of AP-1 dependent transcription. Likewise, additional mutants of this region also showed the ability to transrepress both AP-1 and NF-κB dependent transcription and were again unable to activate GRE dependent transcription.62 ,64 ,78
The above studies have clearly shown that some of the repressive and activating functions of GR may be dissociated at the protein level. The question that then arises is whether these functions can be differentially activated by steroid ligands. From a therapeutic point of view, this could have great significance if, as mentioned above, the repressive functions of GR mediate the anti-inflammatory effects whilst gene activation is responsible for the metabolic, and therefore undesirable, effects of glucocorticoids. Some degree of functional separation is achieved by steroid antagonists such as RU38486 (RU486). This compound shows little ability to transactivate GR dependent transcription.62 ,64 ,79 However, in reporter assays RU486 repressed AP-1 dependent transcription to about 50–70% that for dexamethasone,64 ,79 whilst repression of NF-κB dependent transcription was no more than 30% that for dexamethasone.62 ,78 In addition, the steroids ZK98296 and ZK98299 were identified which showed little ability to transactivate, yet retained 50–80% of the ability of dexamethasone to repress an AP-1 dependent reporter.64Likewise, at 100 nM the glucocorticoids RU24782, RU24858, and RU40066 showed no more than 35% of the ability of dexamethasone to transactivate a GRE dependent reporter, whilst retaining at least 58% of the capacity of dexamethasone to transrepress AP-1 dependent promoters.79 Interestingly, in a monocytic cell line RU24782, RU24858, and RU40066 were found to be poor activators of the glucocorticoid inducible TAT gene but were 70–95% as effective as dexamethasone in suppressing LPS induced release of IL-1β.79 Furthermore, in a cotton pellet granuloma model of inflammation both RU24782 and RU24858 displayed anti-inflammatory properties that were similar to prednisolone, suggesting that transrepressive mechanisms play a significant anti-inflammatory role in this model.79 Similar approaches were also used to dissociate transrepression of NF-κB from GR dependent transactivation.62 In this study the glucocorticoids ZK57740 and ZK077945, which are not anti-inflammatory, showed an impaired ability to transrepress an NF-κB dependent reporter but were fully functional in GRE dependent transactivation assays.62 Conversely, the dissociated glucocorticoids RU24782 and RU24858 were nearly as effective as dexamethasone in the repression of NF-κB dependent transcription, IL-6 promoter activity, and IL-6 expression, whilst remaining non-functional in GRE dependent transactivation assays.57 The above studies suggest that transactivation defective steroids may not induce metabolic genes, and may not therefore give rise to Cushing type symptoms, yet may still illicit useful anti-inflammatory effects. However, other undesirable effects on, for example, bone metabolism may also be mediated via transrepressive mechanisms involving negatively regulated genes such as osteocalcin (fig 1).80 ,81 Indeed, preliminary data suggest that, whilst the dissociated steroid RU24858 showed anti-inflammatory properties in vivo, effects on bone metabolism were similar to glucocorticoids such as budesonide or prednisolone.82 Consequently, whilst the ability of “dissociating” glucocorticoids to separate GR functions can be demonstrated in reporter assays, considerable further work is required to establish whether these compounds can be effectively used in vivo to produce anti-inflammatory effects with reduced side effects.
Gene knockouts and thedim mouse
One powerful tool for studying genetically engineered proteins in an in vivo context is the use of transgenic mice. Mice homozygous for a targeted disruption of the GR gene (GR–/–) die within a few hours of birth due to respiratory failure as a result of severely impaired lung development.83 In addition, these mice show substantially reduced expression of gluconeogenic enzymes such as glucose-6-phosphatase and amino acid catabolising genes such as tyrosine aminotransferase (TAT) and serine dehydrogenase. At the level of the HPA axis, GR–/– mice show 20 times higher levels of ACTH and 2–3 times higher levels of circulating corticosterone. Similarly, CRH expression in the hypothalamus of GR–/–mice was about five times greater than that in wild type mice.84 These effects are consistent with impaired negative feedback inhibition of the HPA axis and thus confirm the role of GR in these processes. To gain further insights into the mechanisms of GR action, a line of transgenic mice was established in which the wild type GR was replaced with a receptor containing the A458T mutation in the D loop (fig 2B).85 As this receptor is unable to homodimerise, it is defective in DNA binding and transcriptional activation from classical GRE sites whilst transrepressive effects via tethering interactions with AP-1 and NF-κB are unaffected.28 ,64 Consequently, mice homozygous for the A458T mutation, referred to as GRdim/dim, offer the opportunity to test in vivo for effects dependent or independent of DNA binding by GR.13 The first significant observation was that, in contrast to GR–/– mice, GRdim/dim mice survive to term and are apparently healthy.85 This immediately implicates functions that are independent of GR DNA binding in normal lung development and other physiological processes (fig 1). Significantly, there was no induction of the TAT gene or other gluconeogenic genes in GRdim/dim mice, indicating that the reporter data were physiologically relevant and that GR dimerisation and DNA binding is necessary in vivo for GR dependent transactivation of these genes. To analyse the effect on tethering mechanisms of transrepression, primary fibroblasts from wild type and GRdim/dim mice were stimulated with phorbol ester and northern analysis performed for the AP-1 dependent genes, collagenase-3, and gelatinase B. In both wild type and GRdim/dim mice a substantial induction of these two genes was observed and in each case this was repressed by 40–45% following treatment with dexamethasone.85 This indicates that the mechanisms involved in the dexamethasone dependent repression of these mRNAs are independent of GR DNA binding (fig 1).
To examine the role of DNA binding in other GR dependent processes, analysis of the HPA axis was also performed in GRdim/dim mice. In the median eminence, which is the site of CRH release into the blood, there was no difference in CRH expression between wild type and GRdim/dim mice, whereas GR–/– mice showed increased CRH expression.83 ,85 In contrast, GRdim/dim mice showed markedly increased mRNA expression of POMC (the ACTH precursor) mRNA in the anterior lobe of the pituitary, whilst ACTH protein expression itself was raised 2.2-fold. Taken together these data indicate that negative feedback control of CRH expression involves effects that are independent of GR DNA binding, whilst mRNA expression of POMC is negatively controlled by mechanisms that are dependent on DNA binding of GR. Furthermore, the fact that POMC mRNA expression was strongly upregulated in GRdim/dim mice, whereas ACTH protein was only increased about twofold, suggests differential control of mRNA and protein expression involving both GR DNA binding dependent and independent mechanisms, respectively. Similarly, expression of prolactin, a gene that is negatively regulated by GR, was also increased in both GR–/– and GRdim/dim mice.85These data suggest that DNA binding dependent effects are involved in the negative regulation of POMC and prolactin and this is consistent with the previous characterisation of nGRE sites in the promoters of these genes.38 ,45
Thus, in vivo analysis of wild type, GR–/–, and GRdim/dim mice confirms the in vitro data by demonstrating separation of DNA binding dependent and independent functions. Furthermore, it appears that transrepression involving tethering mechanisms remains intact, at least in respect of AP-1 dependent genes. Further analyses are now required to examine the ability of these mice to suppress various inflammatory responses in response to glucocorticoids as well as to determine the extent of any other undesirable effects. In particular, it is now important to use these mice to characterise the effect of glucocorticoids on the repression of inflammatory genes, including cytokines and adhesion molecules, that are regulated by transcription factors such as NF-κB, C/EBP, CREB, as well as AP-1. Such studies will shed light on the mechanisms of gene repression and, in particular, the relative contribution of DNA binding dependent and independent effects of GR action. In addition, these studies will act as a validation exercise for the possible therapeutic benefits of second generation dissociating glucocorticoids.
Post-transcriptional and translational targets of glucocorticoid action
So far this review has focused on the transcriptional mechanisms by which glucocorticoids regulate the expression of responsive genes. However, correctly regulated gene expression requires the coordinated control of transcriptional (that is, the rate of gene transcription), post-transcriptional (for example, mRNA stability), translational (that is, protein synthesis), and post-translational (for example, protein processing, modification or degradation) events. In addition, other post-translational processes involving, for example, intracellular localisation or, in the case of cytokines, secretion may also act as control points. Given this myriad of mechanisms involved in the regulation of gene expression, it is likely that a number, if not all, of these processes are also targets of glucocorticoid action.
In recent years it has become increasingly apparent that many genes are regulated to a substantial degree by post-transcriptional and translational mechanisms.86-88 For example, the 3′ untranslated region (UTR) of GM-CSF mRNA contains a number of repeated AU-rich motifs that play a part in mRNA destabilising and translational blockage.89-91 The potential for regulation at this level is illustrated by the fact that proteins binding to these sites can regulate GM-CSF mRNA turnover, whilst mutation of these AU sites enhances GM-CSF expression.92 ,93 In addition, similar AU-rich elements have been described in the 3′ UTRs of numerous cytokine, lymphokine, growth factor, and transcription factor genes, suggesting that similar mechanisms of regulation are, in fact, widespread.94 In this respect, steady state mRNA levels of IL-11 in bone marrow stromal cells were highly inducible by IL-1α and this increase was exclusively attributed to mRNA stabilisation.95 Likewise, post-transcriptional mechanisms have been documented in the induction of numerous inflammatory genes including IL-1β, IL-2, IL-6, IL-8 and COX-2.96-102
As a model system we have studied COX-2 expression and prostaglandin E2 (PGE2) release from human epithelial-like A549 cells. These cells, in common with primary bronchial epithelial cells, produce increased levels of prostaglandins in response to proinflammatory cytokines such as IL-1β and, in both cases, the response is repressed by dexamethasone.103 ,104This induction is at least in part the result of the transcriptional induction of COX-2 by transcription factors such as NF-κB, as well as post-transcriptional mechanisms.101 ,105 In these cells repression of COX-2 expression by dexamethasone was mediated by both transcriptional and post-transcriptional mechanisms.106Dexamethasone reduced the rate of COX-2 transcription by no more than 50% and this correlated closely with the effect on NF-κB dependent transcription in these cells.55 However, as repression of COX-2 protein by dexamethasone was almost absolute, these data clearly implicate the existence of additional repressive mechanisms. In this system dexamethasone decreased COX-2 mRNA stability via mechanisms that involved preferential loss of polyadenylated mRNA.106Interestingly, as repression of mRNA expression by thyroid hormone receptors may also involve the loss of poly(A) tails and the length of the poly(A) tail can have a profound effect on mRNA translation,86 ,107 ,108 these findings could represent a novel mechanism by which nuclear hormone receptors regulate gene expression. In A549 cells this effect required ongoing RNA and protein synthesis, suggesting the need for dexamethasone inducible gene synthesis.106 Furthermore, dexamethasone was highly effective in preventing COX-2 protein expression, even when added a substantial time after the IL-1β stimulus. At these later times, COX-2 mRNA levels are highly increased and inhibition of RNA transcription at these times was ineffective in inhibiting COX-2 expression. These data therefore strongly implicate post-transcriptional and/or translational mechanisms as overriding mechanisms of dexamethasone dependent repression.106 In support of this hypothesis, similar dexamethasone dependent effects have also been reported for COX-2, IL-1β, GM-CSF, IL-6, IL-8, interferon γ, and iNOS in various other cells types.102 ,109-115 In the case of TNF-α, dexamethasone has been shown to act at the level of translation.116 This correlates with the substantial degree of regulatory control that occurs at the level of TNF-α mRNA translation via mechanisms which are thought to involve binding of specific proteins to the AU-rich region within the 3′ UTR as well as by increases in the poly(A) length.108 ,117-119 Furthermore, the suggestion that glucocorticoids act more globally at the level of translation is also supported by the finding that dexamethasone can suppress the synthesis of many ribosomal proteins and translation initiation factors.120 Consistent with the above findings, we have previously found that dexamethasone was ineffective in repressing the activation of GM-CSF promoter constructs in human T cells.121 As GM-CSF release was totally inhibited, these experiments again implicate a significant role for post-transcriptional or translational mechanisms of action. One further example of post-transcriptional downregulation by dexamethasone has been shown for monocyte chemoattractant protein 1 (MCP-1).122 In this case the dexamethasone dependent destabilising activity was found not to require new protein or RNA synthesis. Taken together, these findings indicate that post-transcriptional and/or translational mechanisms of control represent potent glucocorticoid effector functions. Furthermore, the dependence on ongoing transcription or translation indicates that many of these effects involve transcriptional activation and therefore presumably the transactivation functions of GR.102 ,106 ,112 A model for glucocorticoid dependent repression of inflammatory genes is therefore proposed involving both the transrepressive and transactivating functions of GR (fig4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A model for glucocorticoid dependent repression of proinflammatory genes. A generalised inflammatory cascade is shown. Cytokine binding to its cognate receptor localised in the plasma membrane (pm) leads to activation of a kinase cascade consisting of kinases 1, 2, and 3 (K1, K2, and K3). K3 translocates across the nuclear membrane (nm) and then phosphorylates the transcription factor (TF) which actives transcription of an inflammatory protein gene. This leads to mRNA synthesis (transcription) and protein synthesis (translation) of the inflammatory protein. Binding of glucocorticoid to the glucocorticoid receptor (GE) leads to dissociation of the heat shock proteins (hsp90) and translocation of GR to the nucleus. (i) GR may bind TF (for example NF-κB or AP-1) to repress activated transcription via tethering type interactions. (ii) Alternatively, it is hypothesised that GR interacts with other factors (X, Y) to activate gene transcription of anti-inflammatory genes. These anti-inflammatory genes are further hypothesised to promote mRNA degradation and/or repress translation of inflammatory genes.
Positive transcriptional effects of glucocorticoids in immunosuppression
It was noted above that glucocorticoids induce apoptosis of both T cells and eosinophils. In this respect entry of the cell to the programmed cell death pathway is an active process and requires a variety of newly synthesised proteins.123 Likewise, dexamethasone dependent protein synthesis is implicated in cell death and the DNA fragmentation that precedes cell death in T cell apoptosis that has been induced by dexamethasone.123-126 This role for dexamethasone dependent gene synthesis is further substantiated by the finding that thymocytes from both homozygous GR–/–and GRdim/dim mice are resistant to dexamethasone induced apoptosis.83 ,85
Another prominent effect of glucocorticoids on T cells and other cells is the arrest of proliferation by blocking cell cycle progression at G0/G1. Unlike apoptosis, inhibition of T cell proliferation by glucocorticoids occurs at least in part by repression of cell cycle genes such as the G1 progression factor, cyclin D3.127 Remarkably, this repression of cyclin D3 occurs via a rapid post-transcriptional repression mechanism which, like many of the inflammatory genes (above), appears to require the synthesis of a glucocorticoid induced protein or proteins.127 Thus, once again, detailed molecular analysis reveals strong evidence for substantive immunosuppressive effects of glucocorticoids being mediated via positive transcriptional mechanisms.
Other transcriptional responses mediated by glucocorticoids
In the preceding sections we have seen how glucocorticoids, acting via GR, can positively regulate transcription from GRE sites and negatively regulate transcription via a variety of mechanisms including nGRE and tethering sites. However, a number of other transcription modulating activities of GR exist which may result in biologically significant effects. One area that is frequently overlooked is the ability of GR to act synergistically with AP-1 or the AP-1 components c-Fos and c-Jun from composite elements (fig 3B).128 ,129This type of “composite” DNA binding element contains binding sites for GR as well as factors such as AP-1 (fig 3B). Furthermore, the functional outcome of factors binding to these sites depends very much on the cellular environment. Thus, dexamethasone may activate or repress transcription from these elements depending on the cell type.128 Physiologically, these effects resemble that for genes such as PEPCK, which may be upregulated or downregulated by glucocorticoids depending on the cell type.128 Similarly, paired simple GRE and AP-1 sites can result in synergistic activation of transcriptional responses in a manner that is dependent on the spacing between the two elements.130 Likewise, similar cooperative effects have been shown between GRE sites and other transcription factor binding sites.131-133 One group of factors that is involved in the activation of glucocorticoid dependent transcription is the CCAAT/enhancer binding proteins (C/EBP).132 Indeed, C/EBP elements are strongly implicated in the glucocorticoid induction of genes such as the rat arginase gene or the PEPCK gene via mechanisms that involve induction of C/EBPβ.11 ,134 In addition, activation of the PEPCK gene by glucocorticoids and cAMP has also been proposed to involve interaction between GR and CREB.135 Likewise, the synergistic induction of the rat serine protease inhibitor-3 gene by IL-6 and dexamethasone appears to involve interaction of GR with Stat3 and C/EBP.136 Similarly, the transcription factor Stat5 is capable of synergy with GR in a manner that involves direct interaction between Stat5 and GR and does not seem to require GR DNA binding (fig3C).137 ,138
The existence of the above interactions raises the possibility that, in addition to effects in respect of acute phase genes in the liver, these types of response may also play a role in the anti-inflammatory actions of glucocorticoids. At present these interactions have not been examined using dissociated steroids or the various transactivation and transrepression defective GR mutants. Consequently, we do not currently understand the mechanisms behind these effects nor can we guess the role that these processes may play in the anti-inflammatory actions of glucocorticoids. However, it is tempting to speculate that putative glucocorticoid inducible genes, which are involved in the post-transcriptional repression of inflammatory genes, may display similar responses.
Future directions
In recent years a number of glucocorticoid inducible signalling proteins such as the serum and glucocorticoid inducible kinasesgk,25 the glucocorticoid induced diacylglycerol kinase,139 and the small Ras-like GTPase, dexras 1 140 have been identified. These and other similar proteins may be expected to elicit direct, albeit as yet uncharacterised, dexamethasone dependent signalling effects. Furthermore, it is likely that additional glucocorticoid inducible genes involved in signal transduction will be characterised and collectively these new signalling proteins will lead to novel glucocorticoid dependent responses. Two events that are dramatically altering our ability to characterise changes in gene expression at the mRNA level are the revolution in microarray or gene chip technology and the imminent completion of the human genome project. In this respect the sequencing of human chromosome 22 was recently reported.141 When complete, the human genome project, in conjunction with the refinement of current expressed sequence tag site (EST) data bases, will provide investigators with a virtually complete list of human genes. This information can then be combined with array or chip technology to analyse thousands, tens of thousands or, ultimately, hundreds of thousand of genes for changes in mRNA expression following a given stimulus or in a given cell type.142 Thus, the full RNA characterisation, in terms of both positive and negative changes in gene expression, will be possible following glucocorticoid treatment. Such analyses may allow the molecular characterisation of individuals who fail to respond to steroids and could lead to individual specific therapeutic approaches. In addition, the identification of genes involved in the glucocorticoid response will facilitate the identification of responses that are desirable for anti-inflammatory effects and those that are undesirable—for example, those that promote bone metabolism or give rise to Cushing's syndrome.
In conclusion, many of the transcriptional effects of glucocorticoids mediated by GR, including upregulation of gene transcription via “classical” GRE sites and repression of transcription via nGRE and tethering type mechanisms, are currently well characterised. These mechanisms account for a number of positive effects of glucocorticoids including various metabolic effects, as well as certain anti-inflammatory effects—for example, transrepression via interactions with transcription factors such as AP-1 and NF-κB. However, these mechanisms alone do not appear to explain the full ability of glucocorticoids to repress many inflammatory genes. In this respect, an increasing number of studies have documented the existence of post-transcriptional mechanisms of repression that appear to play a significant, if not over-riding, role in glucocorticoid dependent gene repression (fig 4). Furthermore, positive transcriptional mechanisms have been strongly implicated in a number of immunosuppressive effects of glucocorticoids, including post-transcriptional repression, and it is possible that these may be mediated via novel interactions between GR and transcriptional activators (fig 4). The further characterisation of these positive (and negative) transcriptional effects will be greatly facilitated by microarray expression analysis and these strategies can be expected to result in the definition, in terms of changes in gene expression, of the desirable anti-inflammatory properties of glucocorticoids. Finally, these approaches combined with a continued mutational analysis of GR may eventually lead to the characterisation of new classes of glucocorticoids that retain potent anti-inflammatory effects whilst minimising unwanted effects.
Acknowledgments
The author wishes to thank Donna Slater, Karl Staples, and John Matthews for their critical evaluation of the manuscript.