Article Text

Statistics from Altmetric.com
In the early 1950s Lichtenstein introduced the term histiocytosis X to describe a group of syndromes with seemingly unrelated clinical features, but whose pathological findings were characterised by the infiltration of involved tissues with large numbers of unusual histiocytes, often organised as granulomas.1 Subsequently it was shown that these histiocytes were similar to Langerhans' cells normally present in the skin and other epithelia, a finding that led to the current designation of Langerhans' cell histiocytosis (LCH) for this group of disorders.2 ,3
Although the different forms of LCH are united by a common histopathological finding—the Langerhans' cell granuloma—the clinical spectrum of these disorders is extremely large, extending from an acute disseminated form which usually occurs in infants and carries a poor prognosis, to the presence of lesions localised to a single tissue which follows a more benign clinical course (table1).4-8 Pulmonary involvement is not unusual in systemic forms of LCH.4-8 In this setting prognosis depends on the age of the patients, the number of organs involved, and the extent to which normal function is impaired.4-9 Pulmonary symptoms are rarely prominent, although the presence or absence of lung involvement does appear to have a bearing on the outcome.7 ,9 The lung can also be the predominant or only site involved in LCH.4-6 ,8 ,10 Localised pulmonary LCH, the form most frequently encountered by specialists in pulmonary medicine, occurs most commonly in young adults and has several unique clinical and epidemiological features that justify its classification as a distinct clinicopathological entity.4 ,6 ,8 ,11-17The development of high resolution computed tomographic (HRCT) scanning has proved to be of considerable help in the diagnosis of this disorder.18 ,19 In contrast, the natural history of localised pulmonary LCH remains poorly defined and no treatment has been found to be efficacious. Recent progress in the understanding of the pathogenesis of LCH, however, may ultimately lead to the development of new therapeutic approaches.
Clinical spectrum of Langerhans' cell histiocytosis
Epidemiology
Pulmonary LCH in adults is a rare disease and no precise epidemiological data are available concerning its prevalence. In a large series of patients undergoing open lung biopsy for interstitial lung disease of unknown origin, less than 5% were diagnosed as having pulmonary LCH.20 Because the disease can be asymptomatic, undergoes spontaneous remission in a substantial portion of patients, and may be difficult to identify from lung biopsy specimens in patients with very advanced disease, it is probably underdiagnosed. The increased use of HRCT scanning in the evaluation of patients (see below) may lead to an increase in the number of patients in whom the diagnosis is established.
Pulmonary involvement with LCH can be observed in patients of any age.4-6 ,8 ,10 Multifocal and systemic forms of the disease are usually seen in infants and children and pulmonary involvement is often not a prominent feature. In contrast, isolated pulmonary LCH occurs predominantly in young adults with a peak frequency between 20 and 40 years of age.4 ,6 ,8 ,11-17The reported sex ratio has varied considerably in different studies. Some have reported a clear male predominance11 ,16 while others found a nearly equal male to female ratio.14 ,17Several American series limited to patients for whom biopsy specimens were available found a higher incidence in women.8 ,12 ,13 ,15 In one of these series the age of onset was higher in women than in men.12 These variations may reflect differences in the smoking habits of the patients studied. Indeed, the principal epidemiological factor associated with pulmonary LCH is smoking: 90–100% of patients have been current smokers in almost all series.6 ,8 ,11-17 ,21 Patients with pulmonary LCH also tend to be heavy smokers as assessed by daily cigarette consumption but, given the young age of most patients, lifetime cigarette consumption can be relatively modest.21 No correlations between cigarette consumption and the severity of the disease have been found. Little information on racial predisposition is available.
No other clear predisposing factors have been described but an association has been reported between pulmonary LCH and lymphoma.22 ,23 Focal LCH lesions have been identified in lymph node biopsy specimens from areas adjacent to lymphomatous involvement,22 ,23 but clinically apparent pulmonary LCH is more likely to be observed in the follow up of patients previously treated with chemotherapy and/or radiation therapy, usually for Hodgkin's disease.22-24
Clinical presentation
Pulmonary LCH can present in a variety of ways.4 ,6 ,8 ,11-17 Fortuitous discovery on a routine chest radiograph is not infrequent (36% of cases in a recent series).14 Nevertheless, approximately two thirds of patients present with respiratory symptoms, usually a dry cough, often associated with dyspnoea on exertion. These symptoms have generally been persistent (on average six months in a series that provided this information) and may be associated with constitutional symptoms such as fever, weight loss, or asthenia.12 Chest pain may also be the presenting symptom. In a few cases this is caused by rib involvement, but more frequently pain is a sign of a pneumothorax resulting from rupture of a subpleural cystic lesion, a complication seen in approximately 10–20% of patients.8 ,12 ,14-17Pneumothorax can occur at any time throughout the evolution of the disease and can be recurrent or bilateral, but appears to be particularly frequent in younger men.12 ,14-17 ,25 More unusual presentations of pulmonary LCH include wheezing and haemoptysis (in one case due to secondary Aspergillusinfection).11 ,12 ,15 ,17 ,26 Finally, lung involvement can be uncovered during the evaluation of patients presenting with extrathoracic LCH (almost always diabetes insipidus, bone or skin lesions).4 ,6 ,8 ,11-17 Physical examination of the chest is often normal except in patients with pneumothorax, rib lesions or those presenting with advanced disease in whom signs of cor pulmonale are frequently present. Similarly, general physical examination is usually normal unless signs of extrapulmonary lesions are present. Clubbing is an exceptional finding.
Radiological and scintigraphic studies
CHEST RADIOGRAPHS
The abnormalities seen on the chest radiograph depend on the stage of evolution of the process. They are generally bilateral and symmetrical but, although diffuse, are often predominant in the upper and middle lung fields and tend to spare the costophrenic angles.6 ,14 ,18 ,27 In disease of recent onset poorly demarcated micronodular lesions (<5 mm) are characteristic. More commonly, reticulonodular shadows are present in which underlying cystic lesions may be identifiable.6 ,18 ,27 In patients with long standing disease, nodular lesions may be absent and the lung may have a cystic or even a pseudoemphysematous appearance.6 ,18 ,27 Other findings may also be helpful in differentiating pulmonary LCH from other diffuse infiltrative lung diseases, including the sparing or apparent increase in lung volumes, the topographical distribution of the lesions, the absence of mediastinal adenopathy and pleural involvement and, in some patients, the presence of a pneumothorax or of lytic rib lesions. It should be remembered that routine chest radiographs are normal in some patients (<10% of cases).28
HIGH RESOLUTION COMPUTED TOMOGRAPHY (HRCT)
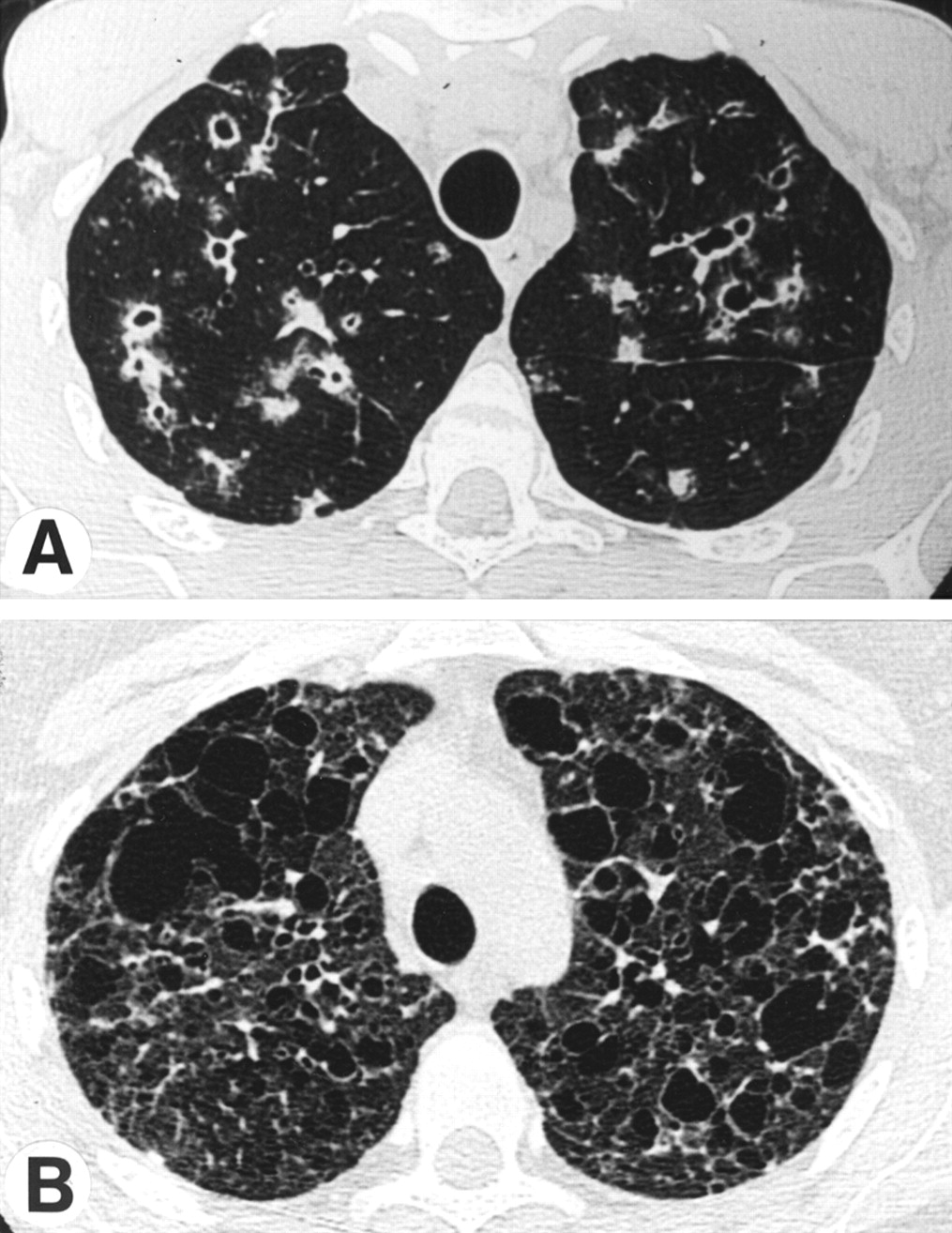
The use of HRCT scanning permits excellent definition of the nature and distribution of parenchymal abnormalities and has proved to be an important advance in the evaluation of patients suspected of having pulmonary LCH.18 ,19 The abnormalities are generally interspersed in parenchyma of normal appearance. The lesions are evenly distributed between the central and peripheral portions of the lung, although the basal portions of the lung tend to be partially spared. In cases of recent onset, small poorly limited nodular lesions predominate, some of which may be cavitary (fig 1A).18 ,19As expected for this bronchocentric process, the nodules have a centrilobular distribution and are usually associated with thin and thick walled cystic lesions.18 ,19 As the disease evolves, cystic lesions become a dominant finding. They are of variable size, usually <1 cm in diameter, and may be isolated, but confluent cysts also form (fig 1B) and, when extensive, give the lung an emphysematous appearance.18 ,19 Serial studies of individual patients indicate that the lesions evolve as follows: nodules, cavitary nodules, thick walled cysts, thin walled cysts.18 ,19 ,29 These studies also show that, whereas some lesions such as nodules and cavitary nodules can regress, cystic lesions tend to remain stable or to increase in size.29

(A) High resolution computed tomographic scan of the lung of a patient with pulmonary LCH showing the association of nodules (some of which are cavitary) and parenchymal cysts. (B) An advanced stage of the disease showing the presence of numerous cysts of various size, some of which are confluent.
The simultaneous presence of nodules, cavitary nodules and cysts is seen in most patients and is highly suggestive of pulmonary LCH.18 ,19 These findings are identified much more reliably on HRCT scans than on standard chest radiographs and can be seen in patients whose chest radiographs are interpreted as normal. Similarly, HRCT scans often show that “reticular” abnormalities on chest radiographs represent the confluence of shadows produced by multiple cystic lesions.19 More unusual findings on HRCT scans include areas with ground glass opacities, linear shadows, and bullous lesions without intervening cystic walls similar to those seen in centrilobular emphysema associated with smoking.29 In one study an increase in the diameter of pulmonary arteries, suggestive of pulmonary arterial hypertension, was observed in five of 17 patients.18 Evidence of pleural fluid and hilar adenopathy is extremely rare.18 ,19 ,30 ,31 In patients who require a lung biopsy HRCT scanning is indispensable for choosing an appropriate site.
SCINTIGRAPHIC STUDIES
Gallium-67 scans are generally negative in pulmonary LCH. Ventilation/perfusion lung scans show only non-specific findings such as non-homogeneous uptake, non-segmental perfusion defects, and air trapping.
Pulmonary function testing
The consequences of pulmonary LCH on pulmonary function are extremely variable and depend on the duration of the disease and the nature of the parenchymal abnormalities present.11-17Furthermore, it is not uncommon to find extensive radiographic abnormalities that appear to be out of proportion to the minimal changes in pulmonary function produced, and in 10–15% of patients resting pulmonary function is normal despite the presence of radiographic abnormalities.11 ,17 Obstructive, restrictive, and mixed patterns have been described.11-17Commonly, the vital capacity (VC) is reduced whereas the residual volume (RV) is normal or increased, resulting in a relatively normal total lung capacity (TLC) and an increased RV/TLC ratio.11 ,14 ,17 This pattern appears to correlate with the presence of extensive cystic changes and/or bullous lesions on the chest radiograph.11 ,14 Evidence of airflow obstruction out of proportion to total cigarette consumption is also common, and approximately half of all patients have abnormalities in the flow/volume curve.32 A frank decrease in the ratio of the forced expiratory volume in one second (FEV1) to forced vital capacity (FVC) is identified in approximately 20% of patients with disease of recent onset, but is more frequent in patients with long standing disease whose chest radiographs show extensive cystic lesions.11 ,14 ,16 ,32 Pure restrictive changes are also encountered.14 ,16 ,17 ,32 Thus, the presence of predominantly obstructive abnormalities in pulmonary function, rarely observed in most diffuse interstitial lung diseases, is a useful indicator for a diagnosis of pulmonary LCH.
The most frequent abnormality in pulmonary function is a decrease in carbon monoxide transfer factor (Tlco) which has been reported in 70–100% of patients in different series.11-17 ,32 Resting blood gas tensions are typically normal or show moderate hypoxaemia, but exercise limitation associated with hypoxaemia is often observed.14 ,17
Fibreoptic bronchoscopy and bronchoalveolar lavage
Visual inspection of the bronchial tree is normal or reveals non-specific inflammatory changes typical of heavy smoking. Similarly, bronchial biopsy specimens are not useful in diagnosing pulmonary LCH and are only indicated if other abnormalities are seen or to exclude alternative diagnostic possibilities. The diagnostic yield from transbronchial biopsy specimens has been variable in published series but is generally reported to be low, perhaps reflecting the focal nature of active lesions.15 ,33 ,34 Nevertheless, the diagnosis can be established from transbronchial biopsy specimens if sufficient material is obtained and specimens are thoroughly evaluated including, if necessary, the use of immunohistochemical techniques to help identify Langerhans' cells.15 Because of the presence of cystic lesions in many patients, however, the risk of pneumothorax is probably higher than usual in these patients.
In most patients the total number of cells recovered by bronchoalveolar lavage (BAL) is considerably increased (up to 106 cells/ml) due to the presence of large numbers of pigment laden macrophages.21 This macrophage alveolitis is not specific for pulmonary LCH but merely reflects the smoking history of the patients.21 Total cell counts correlate with daily cigarette consumption, and increased numbers of macrophages are not found in the small number of non-smokers with the disease.21 Differential cell counts may reveal a moderate increase in the percentage of neutrophils and eosinophils above that seen in smoking control subjects. The proportion of lymphocytes is normal or reduced and the CD4/CD8 ratio is decreased, as in cigarette smokers.21 Langerhans' cells can be identified in BAL fluid by immunocytochemical techniques using anti-CD1a antibodies such as OKT6, and it was initially hoped that this procedure would prove to be a useful non-invasive technique for the diagnosis of pulmonary LCH.35 Further study, however, has put important reservations on the sensitivity of this test for several reasons. Firstly, a number of technical pitfalls can lead to false positive results if the test is not performed by experienced personnel. Secondly, increased numbers of Langerhans' cells are recovered from cigarette smokers and patients with other diseases that lead to alveolar epithelial hyperplasia (including fibrotic interstitial lung diseases) and, in our experience, the number of Langerhans' cells observed in such patients overlaps to a large extent with the number recovered from patients with pulmonary LCH.6 ,36 ,37 Thus, in contrast to some reports,38 ,39 especially those evaluating infants and children,40 we found that, if the threshold for a positive result is fixed at 5% Langerhans' cells, the test is specific but the sensitivity is quite low (<25%).
Laboratory tests
Routine serum biochemical and haematological tests are generally within normal limits. Peripheral blood eosinophilia is not seen in pulmonary LCH. The sedimentation rate can be moderately raised. The presence of low titres of various autoantibodies and circulating immune complexes have been reported but are of no diagnostic significance. Serum angiotensin converting enzyme activity is normal in most patients.
Atypical forms of pulmonary LCH
A few patients may present with unusual clinical features leading to difficulties in establishing the diagnosis. Atypical findings on chest radiographs include the presence of mediastinal lymph nodes, nodules in the absence of cystic lesions, single nodules, markedly asymmetrical findings, cystic lesions with air fluid levels, and localised alveolar consolidation.31 ,41-43 Patients with endotracheal and endobronchial lesions have also been described.41 ,44
Several patients referred for lung transplantation have recently been found to have severe pulmonary arterial hypertension.45The haemodynamic profile observed in these cases is similar to that of patients with primary pulmonary hypertension and can occur in the absence of severe impairment in ventilatory function or hypoxaemia.45
The coexistence of pulmonary LCH with lytic bone lesions, skin lesions, or diabetes insipidus due to extrapulmonary involvement is not infrequent.8 ,11-17 Rarely, adult patients may have pulmonary involvement in the setting of diffuse LCH, a form usually encountered in young children.46
Pathology of pulmonary LCH
The characteristic lesion of pulmonary LCH is composed of activated Langerhans' cells organised into a loose granuloma and associated with lymphocytes and inflammatory cells, particularly eosinophils and macrophages.4 ,6 ,11-13 ,15 The morphology of Langerhans' cells in LCH granulomas is generally similar to that of Langerhans' cells in normal tissues.47 ,48They are of medium size (15 μm diameter) with a poorly demarcated contour because of the presence of multiple cytoplasmic extensions. The nucleus is typically irregular and highly folded, and the cytoplasm is pale, slightly acidophilic, and contains few or no phagocytic vacuoles. At the electron microscopic level the nucleolus is prominent and the cytoplasm contains a well developed Golgi apparatus and abundant endoplasmic reticulum and mitochondria (fig 2A). A characteristic feature of Langerhans' cells seen only by electron microscopy is the presence of membrane demarcated intracytoplasmic organelles—Birbeck granules—the presence of which enables the cells to be unequivocally identified as Langerhans' cells (fig 2B). When cut parallel to their long axis these plate-like organelles have a thickness of 35–45 nm, a pentalaminar structure, and a striated central axis. Their length is variable (100–400 nm); dilatations may be present and, when they occur at the terminal portion of the granule, give the structure a characteristic “tennis racket” shape. Although the Birbeck granules of Langerhans' cells present in LCH granulomas are structurally similar to those seen in normal Langerhans' cells, they are present in much greater numbers49 which probably explains why granules with one end in continuity with the cell membrane are more frequently observed in LCH. The function of Birbeck granules has not been established with certainty, but in vitro studies suggest that they may be involved in the intracytoplasmic transport of antigens captured by Langerhans' cells.50

(A) Electron photomicrograph of a Langerhans' cell with a characteristic folded nucleus. Note the presence of large numbers of mitochondria in the cytoplasm. Original magnification ×7500. (B) Higher power magnification of a Langerhans' cell in an LCH lesion showing the presence of numerous Birbeck granules. Original magnification ×30000.
Langerhans' cells in LCH granulomas, like normal Langerhans' cells, express class I and class II major histocompatibility (MHC) antigens, β2 integrins (CD11a/CD11c/CD18), adhesins (CD54, CD58), leucocyte common antigen (CD45RO), and the intracellular S-100 protein.48 ,51 Certain surface antigens are expressed more strongly by Langerhans' cells in LCH granulomas than by normal Langerhans' cells (table 2).51-54 CD1 molecules are non-polymorphic transmembrane proteins that are structurally related to class I MHC molecules, and may play a role in presenting glycolipid antigens to T cells. Although normal pulmonary Langerhans' cells are CD1a+/CD1c–, those in LCH granulomas strongly express both CD1a and CD1c.49 In addition, Langerhans' cells in LCH can express surface markers associated with activation, such as B7 molecules, that are not present on normal pulmonary Langerhans' cells and that are probably important in the pathogenesis of LCH.55Interestingly, Langerhans' cells that accumulate in other pulmonary diseases such as bronchogenic carcinoma do not show such evidence of activation.51 ,52 ,55
Comparison of the surface phenotype and morphological characteristics of Langerhans' cells present in normal lung, bronchogenic carcinoma, and Langerhans' cell histiocytosis (LCH) granulomas
The appearance of LCH granulomas is partly dependent on the tissue in which they form. In the lung the lesions are focal and are separated by apparently normal lung parenchyma. They are centred around distal bronchioles and infiltrate and destroy the airway walls. In view of this characteristic feature, LCH should be considered to be a bronchiolitis rather than a diffuse interstitial lung disease. The granulomas in pulmonary LCH are poorly defined, however, and extend into adjacent alveolar structures. The surrounding alveoli can contain large numbers of macrophages (a DIP-like reaction) associated with Langerhans' cells. In areas not involved by the granulomas the lung architecture is normal, but non-specific lesions associated with cigarette smoking (respiratory bronchiolitis, accumulation of pigmented macrophages in the alveoli, small lymphoid aggregates in the alveolar walls) are commonly present in these patients.6 ,11-13 ,15
The histology of pulmonary LCH granulomas varies considerably as the lesions evolve, but lesions in different stages of evolution are frequently present in the same biopsy specimen. The earliest lesions form in terminal and respiratory bronchioles and progressively invade and destroy the bronchiolar wall, often extending into the adjoining vascular structures (fig 3). The granulomas contain large numbers of Langerhans' cells forming a compact mass at the centre of the lesion. Lymphocytes are found between the Langerhans' cells and at the periphery of the lesion and are the only other abundant cell type in early lesions. Most of the lymphocytes are CD4+ T cells expressing α/β T cell receptors, many of which also express surface antigens typical of activated cells.49 ,52 A few CD8+ T cells are also present.49 ,52 Inflammatory cells, especially eosinophils and macrophages, occur in variable numbers, usually at the periphery of the granulomas. The destruction of the overlying bronchiolar epithelium is an early event in the pathological process and, as a consequence, it can be difficult to appreciate in a given section the bronchocentric localisation of individual lesions (fig 4). Reconstruction of Langerhans' cell granulomas by analysis of serial sections demonstrates, however, that the lesions follow the involved bronchioles in an uninterrupted fashion, forming a granulomatous sleeve that extends to the respiratory bronchioles of the involved acinus. Early lesions often appear to be excavated. This central cavity does not result from necrosis, but rather represents the lumen of the pre-existing bronchiole destroyed by the granulomatous reaction.
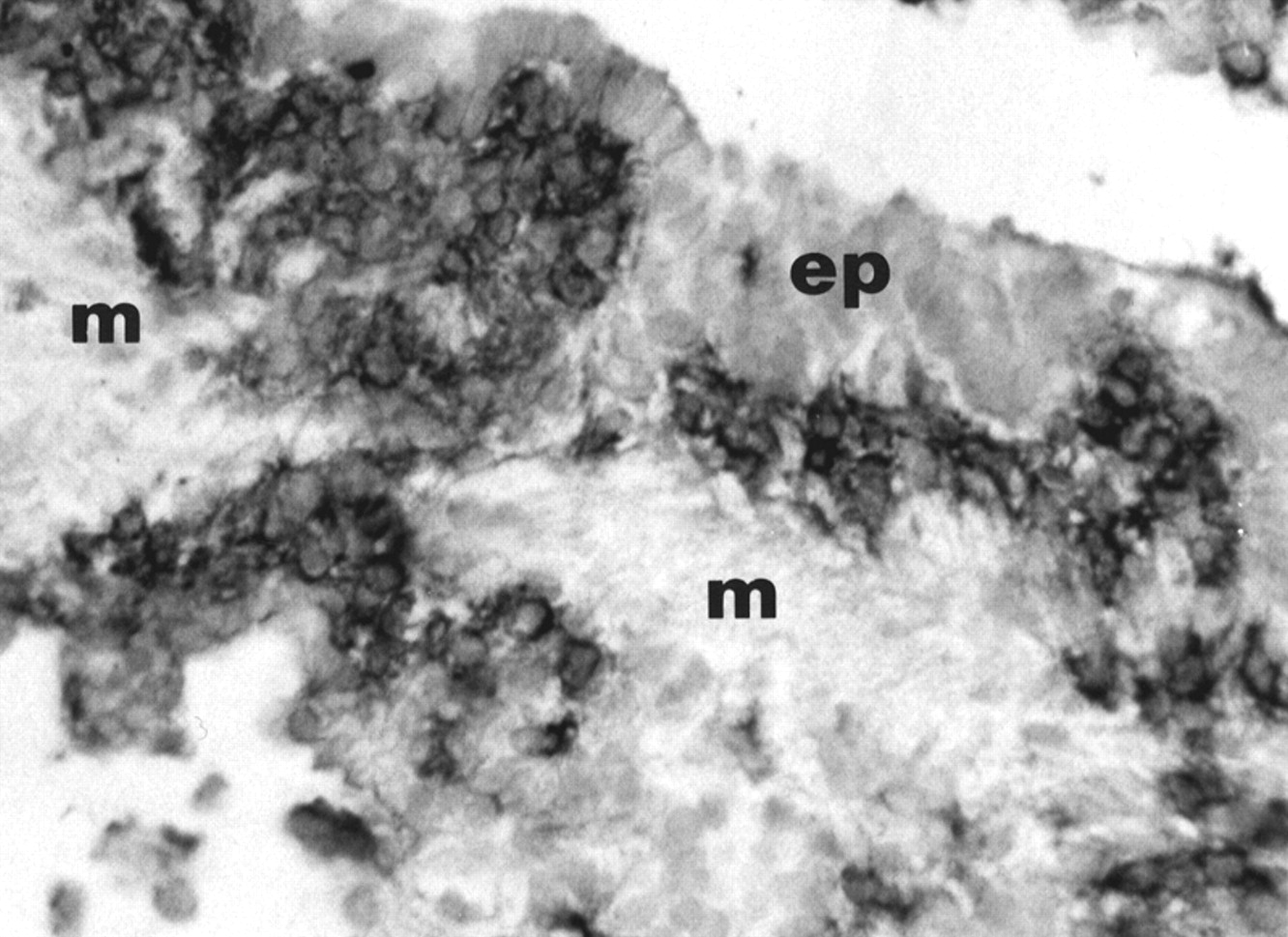
Photomicrograph of an early lesion of pulmonary LCH stained with an anti-CD1a antibody using immunohistochemical techniques. Langerhans' cells accumulate between the bronchiolar epithelium (ep) and the muscle fibres (m) surrounding the bronchiolar mucosa. Note that the cells infiltrate the peribronchiolar tissue. Original magnification ×125.
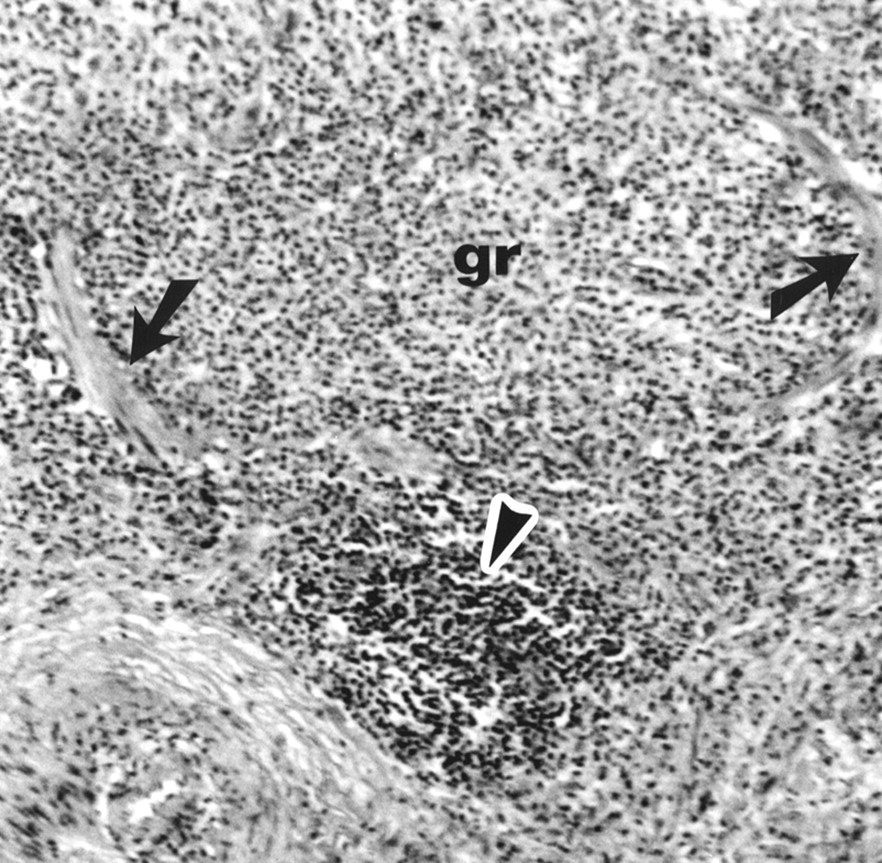
Pulmonary Langerhans' cell granuloma (gr) which has destroyed the bronchiolar architecture. Only remnants of muscle cells (arrows) are recognisable. A lymphoid aggregate is indicated by the arrowhead. Original magnification ×300.
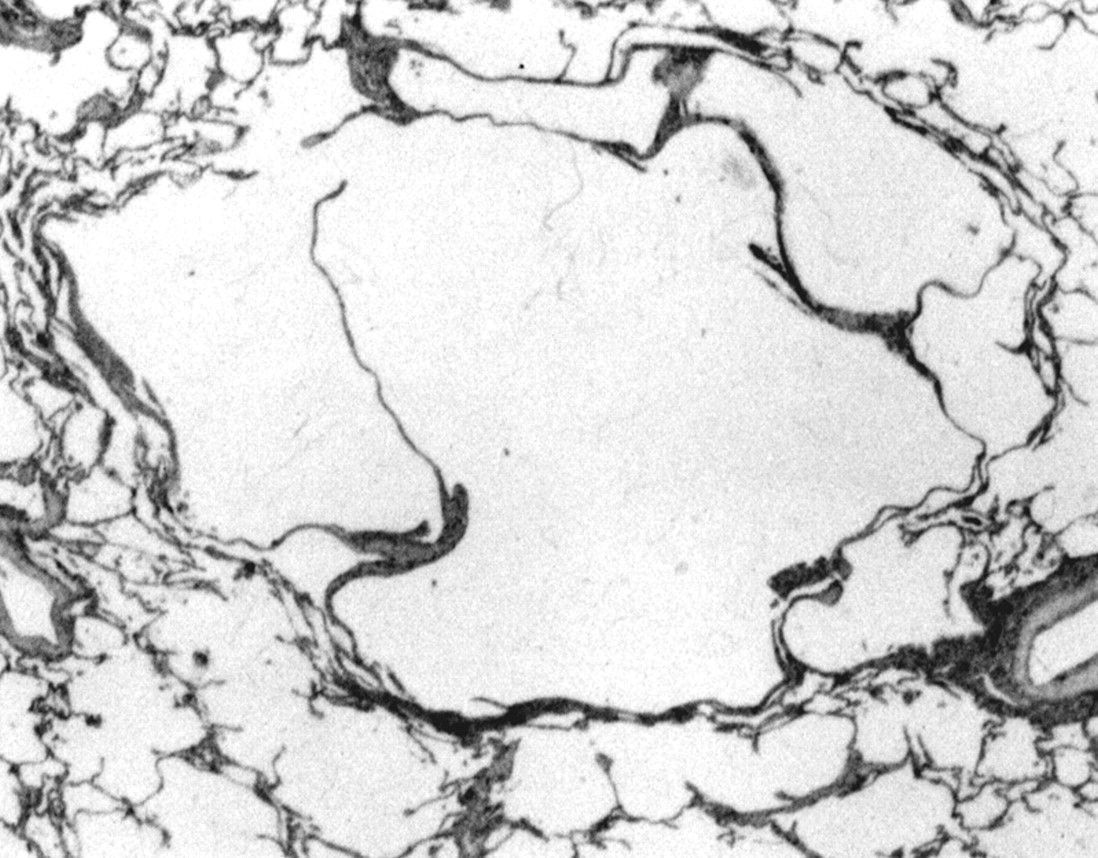
In lesions of intermediate age Langerhans' cells represent a smaller proportion of the total. They are present as small nests of cells surrounded by large numbers of lymphocytes, macrophages, eosinophils, and, to a lesser extent, neutrophils. Lymphoid follicles can be present at the periphery of the lesions, along with evidence of a fibrotic reaction. In late stage lesions Langerhans' cells are present only in small numbers or they may be entirely absent, although pigment laden macrophages and lymphoid follicles persist at the periphery of these late lesions. The lesions heal by fibrosis. In some cases fibrotic rings surrounding cystic cavities of variable size are formed, corresponding to the encasement of the residual dilated lumen of the entirely destroyed bronchiole by the fibrotic reaction (fig 5). In other areas the central lumen is obliterated by characteristic stellate scars. In both settings retraction emphysema of the adjacent alveoli contributes to the cystic appearance of these end stage lesions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Characteristic end stage lesion of pulmonary LCH. Original magnification ×25.
Diagnosis
A firm diagnosis of pulmonary LCH requires the histological demonstration of Langerhans' cell granulomas in the lung tissue. Although the diagnosis can be established using samples obtained by transbronchial lung biopsy,15 open lung biopsy guided by videothoracoscopy is usually preferable to obtain adequate tissue specimens. Several precautions should be observed to optimise the diagnostic yield: (1) the biopsy sites should be shown by HRCT scanning to contain nodular lesions; (2) samples of sufficient size should be obtained; (3) all samples should be thoroughly evaluated, including the use of immunohistochemical techniques to facilitate the identification of Langerhans' cells. In cases where snap frozen samples were not stored at the time the biopsy specimen was taken, the anti-CD1a antibody O10, which recognises the antigen in fixed and embedded tissues, can be used for immunohistochemical studies.56 ,57
In many cases, however, a presumptive diagnosis can be established without tissue confirmation, and the decision to proceed to open lung biopsy must be made on a case by case basis. We generally perform open lung biopsies in patients with pneumothorax who require surgical intervention, in women with diffuse cystic lesions (to distinguish pulmonary LCH from lymphangioleiomytosis), in symptomatic patients with predominantly nodular lesions who are likely to be treated with corticosteroids, and in patients with atypical presentations. In contrast, open lung biopsies are not usually necessary in patients whose HRCT scan shows nodular lesions and cavitary nodules in association with thin and thick walled cysts, especially in individuals with mild or no symptoms who are to be followed without treatment. Similarly, in patients with extensive lung destruction a biopsy may not be warranted because of the excessive surgical risk. In patients with extrathoracic involvement such as bone lesions, biopsy specimens from these sites can be used to support the diagnosis in patients with otherwise typical clinical and radiological features of pulmonary LCH.
The ease of establishing the diagnosis of pulmonary LCH depends on the clinical and radiological findings. In young smokers who present with coexisting nodular and cystic lesions, the diagnosis poses little difficulty. In patients with systemic symptoms, especially when cavitary nodules are prominent or in the rare cases with purely nodular lesions, other possibilities such as mycobacterial or other infections, sarcoidosis, Wegener's disease, cavitary metastatic lesions, alveolar cell carcinoma, septic emboli, or excavated pneumocystosis must be considered.58 In women with purely cystic lesions pulmonary LCH is difficult to distinguish from lymphangioleiomatosis, and the presence of intra-abdominal angiomyolipomas characteristic of the latter disease should be sought by ultrasound or computed tomographic scanning.59-61
Evolution and prognosis
The natural history of pulmonary LCH is quite variable and, in the absence of precise information concerning the factors that influence the evolution, it is difficult to predict for a given patient. Patients are generally followed using clinical parameters, chest radiography (including HRCT scanning when changes on routine chest radiographs are noted), and assessment of pulmonary function, initially at intervals of at least every six months. HRCT scanning has proved to be useful in understanding the evolution of the pathological lesions, but whether serial evaluation with this technique improves patient management remains to be established.
About half of all patients have a favourable clinical course, either spontaneously or while receiving corticosteroids with partial or complete resolution of radiographic abnormalities. Pulmonary function does not necessarily improve in parallel and airway obstruction may remain stable or even increase. Approximately 10–20% of cases follow a rapidly downhill course, often punctuated by repeated pneumothoraces leading to progressive respiratory insufficiency and cor pulmonale. In the remaining patients chest radiographs demonstrate regression or disappearance of nodules which are replaced first by thick walled cystic lesions and later by persisting thin walled cysts or bullae.29 Long term follow up of these patients is required because, even after years of apparent quiescence, lung function can deteriorate and new nodular lesions can occur, suggesting a recrudescence of disease activity.62 ,63
Several factors are associated with a poor prognosis—namely, onset of disease at a very young or old age, persistence of systemic symptoms, recurrent pneumothoraces, the presence of extrathoracic lesions (except for bone lesions which do not appear to modify the prognosis), the presence of diffuse cystic lesions on the chest radiographs, and major abnormalities in pulmonary function at initial assessment (especially reductions in FEV1, FEV1/VC and, to a lesser extent, increases in the RV/TLC ratio).6 ,8 ,11 ,16 ,46None of these criteria is sufficiently reliable to be used to establish a firm prognosis in individual patients.
Pregnancy does not appear to influence the evolution of pulmonary LCH in most cases, although the exacerbation of diabetes insipidus has been reported.64 ,65 Thus, in the absence of accompanying respiratory insufficiency, pulmonary LCH is not a contraindication to pregnancy.
In addition to the association with lymphoma mentioned earlier, bronchogenic carcinoma has been identified with increased frequency in patients with pulmonary LCH.8 ,23 ,66-68 The diagnosis of carcinoma has been made concurrently with or after the diagnosis of pulmonary LCH in all but one case.23 The development of carcinoma at sites of lung scarring and heavy cigarette consumption may contribute to the development of these tumours but, given their central location in many cases and the spectrum of histopathological types observed, this latter factor appears likely to be predominant.23 ,66-68 Other solid tumours have been reported in association with pulmonary LCH, but such occurrences may be merely coincidental.23
Treatment
Because pulmonary LCH is a rare disease that not infrequently resolves or stabilises without treatment, the efficacy of therapeutic intervention is difficult to evaluate.69 ,70 No treatment has so far been confirmed to be useful in this disorder and no double blind therapeutic trials have been reported.
Oral corticosteroids appear to be useful in controlling systemic symptoms and are often used empirically in patients with pulmonary LCH of recent onset—for example, 0.5–1 mg/kg/day prednisone, gradually tapering over 6–12 months. Corticosteroid use was associated with clinical and radiographic improvement in one recent series, but no significant change in respiratory function was observed.14In a retrospective study patients treated with corticosteroids deteriorated more than untreated patients, but selection bias is likely to explain this finding.16 We generally reserve corticosteroids for symptomatic patients with nodular lesions in the hope of accelerating the resolution of the associated granulomatous and inflammatory processes. Apparent improvement in pulmonary arterial hypertension associated with pulmonary LCH by oral corticosteroids has been described in a single case.71 Cytotoxic agents such as vinblastine and etoposide, often used in the treatment of diffuse LCH in children, should be avoided in isolated pulmonary LCH affecting adult patients.
Because of the strong association between cigarette smoking and pulmonary LCH, every effort should be made to ensure that patients abstain from smoking although the influence of cessation on the evolution of established pulmonary LCH has not been evaluated. At least, smoking cessation decreases the risk of bronchogenic carcinoma, coronary artery disease, chronic obstructive lung disease and respiratory infections, conditions that frequently complicate the subsequent course of these patients. Bronchitis and pneumonia are common causes of acute decompensation in patients with pulmonary LCH and early and aggressive treatment of pulmonary infections is important in the management of these patients.
Pneumothoraces are treated by standard techniques such as drainage and pleurodesis. Pleurectomy should be avoided in patients for whom lung transplantation may ultimately be an option.
Lung transplantation has been performed with success in patients with advanced respiratory insufficiency and in a few patients with severe pulmonary arterial hypertension. Recurrence of pulmonary LCH in the transplanted lung appears to occur in a significant proportion of patients.72-74 This complication can be associated with a clinically important deterioration in lung function, but the effect on overall survival remains to be established.
Pathogenesis of pulmonary LCH
As suggested by its name, Langerhans' cells play an essential role in pulmonary LCH and an understanding of the normal function of these cells is helpful in interpreting current information concerning the aetiology of the disorder. Langerhans' cells belong to the family of dendritic cells but can be distinguished from other cells in this lineage by their tissue location, morphological features, surface phenotype, and functional properties. In this section the biology of cells of the dendritic cell/Langerhans' cell (DC/LC) lineage is briefly reviewed before discussing current hypotheses on the pathogenesis of pulmonary LCH.
DENDRITIC CELLS AND LANGERHANS' CELLS
The initiation of an immune response against foreign proteins requires the participation of antigen presenting cells (also called accessory cells).75 These cells internalise and partially degrade antigens, and express the peptide fragments on their surface in association with major histocompatibility complex (MHC) molecules. The cells then migrate to regional lymphoid tissues where they present the processed antigens to T cells. The recognition of an antigen presented by accessory cells is not sufficient to activate resting T cells; the accessory cell must also deliver appropriate costimulatory signals to the responding lymphocyte. In this regard the interaction between B7 molecules (expressed by the accessory cell) and CD28 receptors (expressed by T cells) is thought to be extremely important.76
DC/LC are specialised accessory cells found in many tissues in humans.77 Numerous studies have shown that DC/LC are extremely potent antigen presenting cells—for example, 10–100 times greater lymphocyte proliferation is seen with DC/LC than with equal numbers of macrophages. It is extremely important to recognise, however, that DC/LC do not normally express all the activities required for antigen presentation at the same time. Tissue DC/LC are typically “immature” cells that can capture, process, and display foreign antigens but have little lymphostimulatory activity.77Following internalisation of foreign antigens, however, these cells migrate to regional lymphoid tissues and during this time they lose the capacity to process new antigens and develop the activities necessary for lymphocyte activation.48 ,77-79 In this regard, Langerhans' cells present in normal lung and other peripheral tissues express weak lymphostimulatory activity. Consistent with this finding, Langerhans' cells exhibit Birbeck granules (which may play a role in antigen internalisation) but do not express B7 costimulatory molecules.48 ,55 ,76 ,77 ,80 Following migration to lymphoid tissues, Langerhans' cells mature into lymphostimulatory dendritic cells, a transition that is associated with the loss of Birbeck granules and the appearance of B7 molecules on the cell surface.48 ,76-81 Thus, epithelial Langerhans' cells are “sentinels” that identify antigens against which an immune response may be necessary, but lack the capacity to induce a local immune response. This temporal and spatial separation in the expression of the activities necessary for antigen processing and lymphocyte stimulation is thought to be an important mechanism through which inappropriate immune responses against innocuous antigens are avoided.48 ,77
DC/LC are derived from CD34+ bone marrow precursors; several lineages appear to exist.77 ,82-85 The best characterised lineage shares a common precursor with that giving rise to monocytes and macrophages.77 ,82-84 Cytokines have been identified that promote the in vitro differentiation of this precursor into DC/LC (GM-CSF, IL-4, TNF-α) or monocytes (M-CSF), respectively.77 ,82-84 In addition, treatment of blood monocytes with cytokines (GM-CSF and IL-4) results in the formation of cells with many characteristics of dendritic cells.77 ,86It remains unclear, however, at what point the differentiation of monocytes and dendritic cells diverge in vivo. Although small numbers of dendritic-like cells have been identified in the peripheral circulation,87 ,88 recent evidence suggests that circulating monocytes, under the influence of cell-cell contacts with endothelial cells and/or cytokines produced in the local environment, may give rise to most tissue dendritic cells.86 ,89 ,90Similarly, the relationship between submucosal dendritic cells and Langerhans' cells within the epithelium is not clearly established. Although distinct precursors have been suggested to exist,84 the distribution of these populations in adjacent compartments, the presence of cells with an intermediate phenotype, and the ability of monocytes to differentiate into cells with characteristics of either dendritic or Langerhans' cells depending on the cytokines present, strongly support the idea that pulmonary Langerhans' cells are derived from dendritic cells present in the submucosa.48 ,77 ,89-92 In this context, transforming growth factor (TGF)-β may be important in this transition. This cytokine promotes the differentiation of monocytes into cells with characteristics of Langerhans' cells (CD1a+, presence of Birbeck granules) and, although dendritic cells are present in TGF-β knockout mice, epidermal Langerhans' cells are absent.90 ,93 The production GM-CSF by epithelial cells also appears to be important in the recruitment and/or differentiation of epithelial Langerhans' cells.48 ,77 ,94
In the lung dendritic cells are present in the connective tissue sheath of the bronchovascular tree, the alveolar interstitium, bronchus-associated lymphoid tissue (BALT), and visceral pleura.48 ,91 ,94-99 In the normal lung Langerhans' cells are found almost exclusively within the epithelium of the tracheobronchial tree where their long processes interdigitate between the epithelial cells to form a highly developed network.48 ,91 ,94 ,97 Although few Langerhans' cells are present in the normal alveolar epithelium, these cells do infiltrate certain lung carcinomas and areas of alveolar hyperplasia seen in smokers or patients with interstitial lung diseases.37 ,48 ,51 ,52 ,91 ,94
PULMONARY LCH
Recent studies have established that Langerhans' cells in LCH are of clonal origin, suggesting that abnormalities in these cells play an important role in the pathogenesis of LCH.100-102Pulmonary LCH has a number of characteristic clinical and pathological features, including the initial selective accumulation of large numbers of Langerhans' cells in distal bronchioles, the capacity of the granulomas to destroy the bronchiole, and the extremely strong association of the disease with cigarette smoking.6 Any hypothesis concerning the pathogenesis of pulmonary LCH must therefore explain how abnormalities of the Langerhans' cells result in these unusual features. In this section, current information concerning the mechanisms that result in the accumulation of Langerhans' cells in the early lesions and those responsible for the destructive nature of the granulomatous lesions are summarised. Although discussed separately, it should be emphasised that the two phenomena may be linked. For example, factors important for the attraction of Langerhans' cells in this disease may also modify the functional activities of these cells.
Accumulation of Langerhans' cells in pulmonary LCH
Normal pulmonary Langerhans' cells are present only within the bronchial and bronchiolar epithelia.48 ,91 ,94 ,97Similarly, the granulomatous lesions in pulmonary LCH are highly bronchiolocentric, suggesting that an epithelial microenvironment remains an important determinant for the accumulation of Langerhans' cells in this disease.6 ,11-13 ,15 ,51 Because of the prominent effects of heavy smoking on the bronchiolar epithelium and the strong association between pulmonary LCH and smoking, the possibility that smoking induced changes in the epithelium promote accumulation of Langerhans' cells is attractive. Epithelial cells are able to produce a variety of cytokines, including factors that influence the proliferation, survival, and differentiation of Langerhans' cells.48 ,77 ,94 ,103 In this regard, bronchiolar epithelial cells overlying early LCH granulomas produce greater amounts of GM-CSF than epithelial cells in adjacent uninvolved bronchioles, compatible with the idea that focal abnormalities influence the initiation of the pathological lesions.104 Mediators produced by other airway cells may also be important. For example, neuroendocrine cells, a source of bombesin-like peptides, are substantially increased in the bronchioles of patients with pulmonary LCH.105 The importance of these and other changes in the airway epithelium in the pathogenesis of pulmonary LCH, however, remains to be established. Furthermore, the bronchiolar epithelium is rapidly destroyed in pulmonary LCH which suggests that factors produced by the epithelium are unlikely to contribute to the persistence of Langerhans' cells in these lesions. Several cytokines including GM-CSF are produced by Langerhans' cells in LCH granulomas (see below), and may contribute to the persistence of established lesions through autocrine or paracrine effects.55 ,106-109
Whatever the role of smoking induced epithelial modifications in the initiation of the process, only a very small proportion of smokers develop pulmonary LCH, indicating that other factors must also be required. Recent evidence strongly supports the idea that intrinsic abnormalities in Langerhans' cells are also important for their massive accumulation in early lesions. Studies evaluating the pattern of X chromosome inactivation in Langerhans' cells from women with LCH have shown that the cells are clonal in origin in patients with both diffuse and localised forms of the disease, including pulmonary LCH.100-102 On the basis of this observation it is tempting to consider LCH as a neoplastic disease resulting from a clonal expansion of Langerhans' cells, but with variable clinical expression. Certainly the aggressive course of diffuse LCH (Letterer-Siwe disease) is reminiscent of a malignant process.5 There are, however, a number of clinical and pathological findings that are extremely difficult to reconcile with the idea that pulmonary LCH is a malignant disorder (table 3). Furthermore, Langerhans' cells in pulmonary LCH lesions have a rather low rate of proliferation, considerably less than that of carcinoma cells, indicating that local proliferation makes little contribution to the accumulation of the cells in this disease.110
Clinical and pathological observations that support or controvert the malignant origin of pulmonary Langerhans' cell histiocytosis (LCH)
In this regard it is noteworthy that clonal cell populations, including cells of haematopoietic origin, have been identified in a number of situations in the absence of overt malignancy.111 The mechanisms that lead to the establishment of clonal Langerhans' cells in LCH remain unknown, although the existence of one or more somatic mutations in haematopoietic progenitor cells is a strong possibility. Several examples exist where such putative mutations can influence the behaviour of only one of the several lineages of haematopoietic cells derived from the progenitor.112 ,113 Taken together, current evidence suggests that the appearance of clonal Langerhans' cells is a necessary event in the development of LCH, but that the functional abnormalities of this population are more subtle than overt malignant transformation. For example, mutations resulting in autonomous or enhanced responses to normal cytokine signals could promote clonal expansion of progenitors. Following additional differentiation, however, these Langerhans' cells could lose the capacity to replicate but continue to have abnormalities that become clinically manifest only under certain conditions—for example, abnormal responses to chemotactic signals, reduced sensitivity to apoptosis, or precocious expression of lymphostimulatory activity. Better understanding of the molecular basis of the clonality of Langerhans' cells will undoubtedly provide important insights into the pathogenesis of the disease.
Mechanisms underlying the destructive nature of LCH granulomas
Regardless of the mechanisms responsible for the accumulation of Langerhans' cells in early LCH lesions, the capacity of the resulting lesions to destroy the bronchiolar structures requires explanation. In other situations in which an abnormal accumulation of Langerhans' cells occurs—for example, at sites of hyperplastic alveolar epithelium or within certain bronchogenic carcinomas—this infiltration is not accompanied by tissue destruction or the recruitment of other inflammatory cells.47 ,51 ,52 ,91
As discussed earlier, Langerhans' cells in LCH granulomas, unlike those present in the normal lung or in other pathological conditions, exhibit a number of phenotypic features characteristic of “mature” Langerhans' cells.51-55 Although not described in other contexts, it is possible that these cells express activities that directly result in local tissue damage. It appears more likely, however, that these “mature” Langerhans' cells express enhanced lymphostimulatory activity and initiate an uncontrolled immune response that damages the airways and adjacent lung parenchyma. Several arguments support this possibility:
- (1)
- The principal function described for Langerhans' cells is to serve as accessory cells in the initiation of immune responses.77 They are now recognised to be extremely important in the development of immune responses in the lung.48
- (2)
- In addition to Langerhans' cells, lymphocytes are the only other cell type present in large numbers in early LCH lesions. As would be expected in the course of an immune response against exogenous antigens, these lymphocytes are predominantly CD4+ T cells with α/β T cell receptors and express a number of markers typical of recently activated cells.49,52 Indeed, examination of Langerhans' cell granulomas at the electron microscopic level has consistently demonstrated the presence of close differentiated contacts between Langerhans' cells and the T lymphocytes in LCH granulomas, including rosette-like clusters of T cells surrounding a central Langerhans' cell, features also seen in cutaneous hypersensitivity reactions.49,114
- (3)
- The structure of the early stage LCH lesions, composed of a central accumulation of Langerhans' cells surrounded by and infiltrated with T cells, is typical of an immune granuloma.
- (4)
- The evolution of the pathological lesions in pulmonary LCH is reminiscent of a granulomatous response. The initial accumulation of accessory cells (Langerhans' cells) and T cells, the subsequent recruitment of inflammatory cells, and the progressive replacement of these cells by macrophages, fibroblasts, and fibrotic tissue also occurs in the course of other granulomatous reactions. As would be expected for an ongoing immune response, local production of numerous cytokines by LCH granulomas has been detected, including cytokines that could contribute to the accumulation of eosinophils in florid lesions (IL-4, IL-5, GM-CSF) and mediators such as TGF-β and metalloproteinases that could contribute to tissue remodelling and the fibrotic response.55,106-109,115
Because tissue Langerhans' cells, including those in the lung, are normally “immature” cells with poor lymphostimulatory activity, it was not initially clear how these cells in LCH lesions could initiate an immune response.48 However, more recent studies have shown that, unlike Langerhans' cells in the normal bronchial mucosa or those that accumulate within lung carcinomas or at sites of alveolar hyperplasia, the Langerhans' cells present in the granulomatous lesions strongly express the costimulatory molecules B7-1 and B7-2 as well as other phenotypic markers characteristic of “mature” cells with strong lymphostimulatory activity.55 The reasons why Langerhans' cells in LCH granulomas express this unusual phenotype are not fully understood. The profile of cytokines expressed in these lesions (presence of GM-CSF, TNF-α, IL-1β, absence of IL-10) correspond to a cytokine profile that has been shown by in vitro studies to induce the maturation of Langerhans' cells into cells with strong lymphostimulatory activity and may explain this unusual phenotype.55 ,76 ,77 ,116-118 Interactions with T cells may also facilitate the expression of lymphostimulatory activity by Langerhans' cells. Langerhans' cells in LCH granulomas strongly express CD40 and many activated T cells present in LCH granulomas, unlike lymphocytes present in the normal lung, express CD154 (CD40 ligand).55 Because CD40/CD154 interactions upregulate the expression of B7 costimulatory molecules on the surface of Langerhans' cells, such a positive feedback loop could contribute to the ability of these cells to initiate local immune reactions.119Finally, intrinsic abnormalities in the clonal population of Langerhans' cells that constitute the lesions may favour the development of cells that express lymphostimulatory activity within the lung, either directly or by increasing the sensitivity of these cells to the differentiating effects of cytokines or other mediators present in the local milieu.
The idea that pulmonary LCH results from the accumulation of abnormal “lymphostimulatory” Langerhans' cells that initiate an uncontrolled immune response presupposes the presence of an antigenic trigger, but such putative antigens remain to be identified. The possibility of a viral aetiology has been raised and a role for a herpes virus (HSV6) has been suggested.120 Recent studies evaluating a large number of candidates have failed, however, to identify viral DNA in pulmonary LCH granulomas.102 The bronchiolocentric distribution of the lesions and the strong association with cigarette smoking has raised the possibility that an immune reaction against a component of cigarette smoke is involved.121 This hypothesis does not explain the occurrence of the disease in children or in the rare non-smoking patients. We have suggested that abnormal bronchiolar epithelial cells could be a target.6 According to this scenario, hyperplastic or dysplastic epithelial lesions would participate in two steps of the process—the production of mediators necessary for the recruitment and/or differentiation of abnormal Langerhans' cells and the expression of neoantigens that are a trigger for the ensuing granulomatous response. The targeting of epithelial cells would account for the bronchocentric nature of the lesions and the early destruction of the epithelium, and would also explain why cigarette smoking, a common cause of epithelial abnormalities, is frequently associated with pulmonary LCH even though non-smokers, who may develop epithelial abnormalities for other reasons (such as radiotherapy), can also develop the disease. Nevertheless, no direct evidence for this possibility has been presented.
In summary, the demonstration that Langerhans' cells in LCH lesions are of clonal origin represents a major advance in our understanding of this disease. Considerable work remains, however, to define how the abnormalities that result in the appearance of these clonal cells change their intrinsic functional properties or modify their response to environmental influences such as epithelial abnormalities or cigarette smoke. Of particular importance will be efforts to define better the role of immune responses initiated by pulmonary Langerhans' cells expressing abnormal lymphostimulatory activity in the pathogenesis of this enigmatic disease.