Article Text

Statistics from Altmetric.com
There have been a number of major advances in molecular biology in the past few years and the aim of this review is to describe some of these advances, focusing on their benefits and limitations when applied to investigating pulmonary disorders. It is written with the practising pulmonary researcher in mind, not as an introduction for the uninitiated. Useful web addresses and a list of references are included to enable interested readers to examine each technique in detail.
New technologies either improve existing techniques or develop new approaches to old questions in order to generate information more quickly, easily, accurately or in a more easily repeatable fashion then existing methods.1 Some of the most powerful new technologies include polymerase chain reaction (PCR) advances, “difference analysis” (that is, the discovery of different gene expression patterns between different cells), transgenic/gene knockout technology, and gene delivery to tissues/gene therapy.
Advances in PCR technology
Since its introduction in the 1980s PCR has become a standard tool in biomedical research. The equipment (a thermal cycler) and reagents (thermostable polymerases, oligonucleotides, etc) required for PCR are widely available and relatively inexpensive. One advantage of PCR is its extreme sensitivity which makes possible the detection and analysis of low abundance DNAs. This is especially helpful when limited amounts of starting material are available, or when few copies of the target sequence are present. Applications of PCR include the cloning of known and novel genomic DNA and cDNA sequences, DNA sequencing, construction of mutant or chimeric DNAs, and quantification of mRNA and DNA. PCR is also used in certain methods of difference analysis
PCR BASED CLONING AND SEQUENCING METHODS
PCR has been used to facilitate cloning of known DNAs and to allow for the identification of novel DNAs. It can be performed directly on genomic DNA and on cDNA produced from mRNA (reverse transcription PCR (RT-PCR)). This eliminates the requirement for the production and screening of DNA libraries. When the target DNA sequence is known, the target DNA can easily be amplified using oligonucleotide primers based on the sequence. When only a portion of a target cDNA sequence is known, PCR can be used to amplify unknown sequences at the 5′ or 3′ ends of the known region (rapid amplification of cDNA ends (RACE)).2 Novel homologues of known proteins can also be identified using a method known as homology PCR.3 This method relies upon the use of degenerate mixtures of oligonucleotide primers designed to recognise conserved motifs. Homology PCR can be used to clone novel members of protein families or to clone protein homologues in other species.
PCR based techniques such as allele specific PCR and PCR restriction length polymorphism (PCR-RFLP) can be used to speed the detection of genetic polymorphisms in large populations. PCR fragments can also be sequenced directly, enabling screening of populations for novel mutations. PCR has been incorporated directly into DNA sequencing technologies, making possible the sequencing of small (nanogram) amounts of target DNA and sequencing of unpurified DNA—for example, for direct sequencing of DNA from bacterial colonies or phage plaques.4
CONSTRUCTION OF MUTANT AND CHIMERIC DNA USING PCR
Mutant and chimeric DNA sequences are often useful for analyses of the structure and function of genes and proteins. Mutations can be introduced by using PCR primers that incorporate one or more point mutations, deletions, or insertions. Two or more distinct DNA sequences can be joined using splice overlap extension PCR.5 This method, which does not require the presence of restriction sites of other specific sequences, greatly facilitates the production of cDNAs encoding chimeric and fusion proteins.
QUANTIFICATION OF mRNA USING PCR
PCR has been widely used to measure the level of expression of mRNAs. The major advantage of PCR based methods of mRNA quantification is high sensitivity. PCR based methods may be the only option if the amount of starting material is limited (as often occurs when analysing mRNA expression in primary cells) or if the target sequence is expressed at very low copy number. However, when RT-PCR is carried out in standard fashion (reverse transcription of RNA followed by a fixed number of cycles of cDNA amplification), the amount of product DNA does not relate in a consistent or predictable manner to the amount of input RNA. There are several reasons for this. For example, there may be contaminants in the RNA preparation that interfere with reverse transcription or amplification, and oligonucleotides will differ greatly in their ability to specifically prime product synthesis. Another major problem is that the efficiency of amplification varies from cycle to cycle. In theory, the amount of PCR product should double each cycle, but in practice this is not the case and the efficiency of amplification drops considerably in later cycles. Two approaches have been widely used to make RT-PCR a quantitative technique: competitor PCR and real time quantitative PCR.
In competitor PCR a competitor DNA construct is added to each PCR reaction (fig 1). The competitor construct is identical to the authentic sequence except for the addition (or deletion) of a portion of the target sequence located somewhere between the two amplification primers. Amplification results in two products—an authentic cDNA product and a competitor product that is somewhat larger (or smaller) than the authentic product. Since the efficiency of amplification of both products should be similar in each cycle, the ratio of authentic product to competitor provides a good quantitative (or at least semiquantitative) estimate of the amount of input cDNA. Expression of specific mRNAs can be normalised by comparison with one or more “housekeeping” gene. Competitors are available for an assortment of genes and can be readily produced by various methods.6 ,7The method is well suited for analysis of the expression of a small to moderate number of mRNAs. For example, competitor PCR was used to analyse the expression of cytokine mRNAs in a murine model of asthma (fig 2).8 Careful titration of input cDNAs and competitors may be required, especially to produce more rigorous quantification, and the method may be quite labour intensive, especially when large numbers of samples are to be analysed.
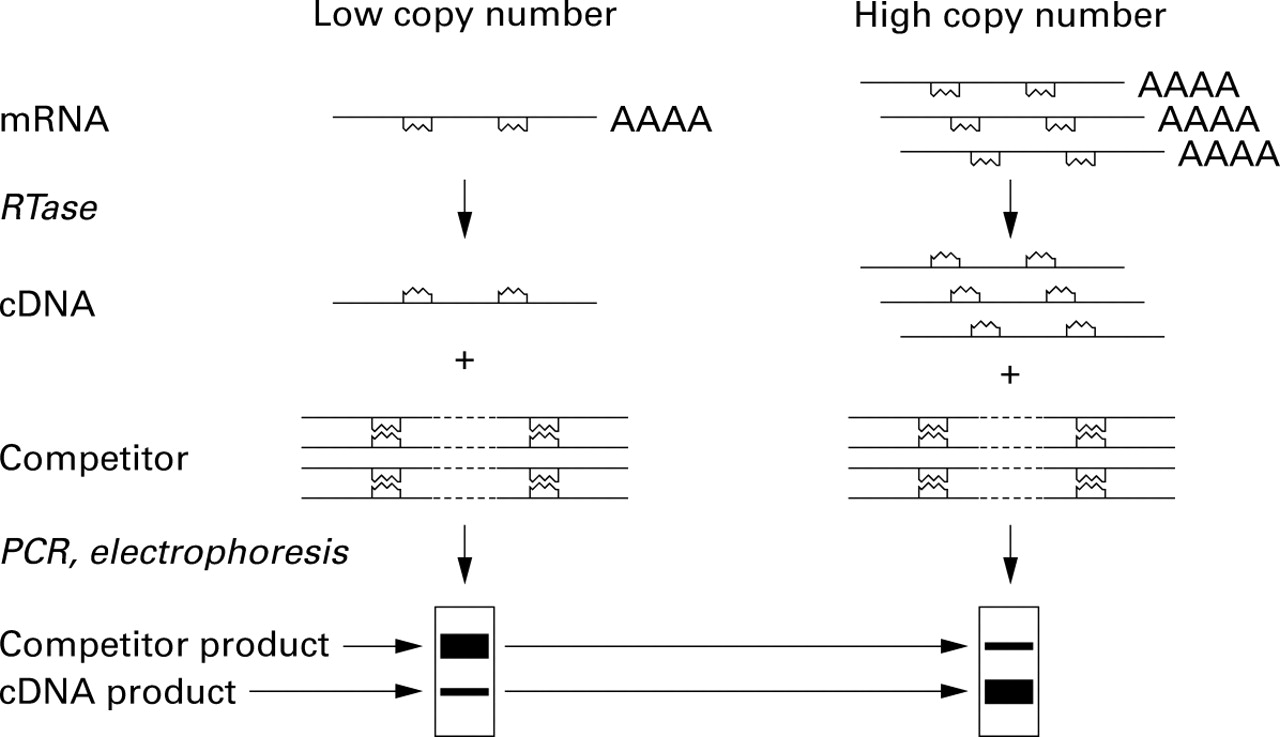
Competitor PCR. Reverse transcriptase (RTase) is used to synthesise cDNA from RNA. A known amount of a competitor DNA construct is then added to each cDNA and PCR is performed. In the case shown here the competitor has been prepared by insertion of an arbitrary short sequence of DNA into the region that will be amplified by the primers, resulting in a larger PCR product. When a sample containing few copies of the mRNA is analysed the competitor product is more abundant than the competitor. When the sample contains many copies of the mRNA the smaller authentic product dominates. By normalisation of starting material (using competitor PCR to analyse expression of one or more “housekeeping” genes) and careful titration of competitors, it is possible to achieve sensitive and reasonably quantitative measurements of mRNAs.

Semiquantitative analysis of cytokine mRNA expression. Bronchoalveolar lavage fluid leucocytes from a murine asthma model were obtained from mice that were immunised and then challenged with aerosolised ovalbumin (OVA, lane 3) and control mice that were only immunised (lane 1) or challenged (lane 2). After cDNA synthesis the amount of input cDNA was normalised using a housekeeping gene (hypoxanthine phosphoribosyl transferase, HPRT). Note that the relative intensities of the authentic (lower) and competitor (upper) HPRT bands are similar in all three lanes. In contrast, levels of cytokine mRNAs are substantially different in the three conditions. For example, IL-4 and IL-5 expression is increased dramatically in ovalbumin immunised and challenged mice.
A second approach to quantitating mRNAs is real time quantitative PCR. This method involves the use of a specialised thermal cycler capable of measuring fluorescence in each reaction during each cycle. A fluorescent signal is produced either by using a dye (SYBR Green) that fluoresces after binding to double stranded DNA, or by using a special oligonucleotide with a fluorescent reporter dye that is released by the DNA polymerase during each extension cycle. The amount of input cDNA is quantified by identifying the “threshold cycle”, the first cycle in which product can be unambiguously detected. Lower threshold cycle numbers are associated with higher amounts of input cDNA. Since PCR products are detected early, problems attributable to non-exponential amplification are minimised. This method can be made highly reproducible and is especially well suited for the measurement of one or a few mRNAs from a large number of sources, as may be required for screening the effects of libraries of compounds. It does, however, require a relatively expensive specialised cycler and careful development and testing of appropriate oligonucleotide primers for each mRNA.
Difference analysis
Finding genes/proteins that are differentially expressed in diseased tissues or altered cells is a key to understanding the pathogenesis, aetiology, and/or response of diseases to treatment. Difference analysis is more readily achieved by examining mRNA differences than protein differences, largely for technical reasons—it is substantially easier to identify differentialgene expression thanprotein expression in disease tissues. The early approaches to analysis of differential mRNA expression relied upon subtractive hybridisation, an approach that has detected the expression of a number of important genes despite the time consuming and labourious nature of this approach. It does not detect mRNAs which are expressed at very low levels. As there are up to 100 000 human genes and a single cell expresses perhaps 25 000 of these as distinct mRNA transcripts, with 98–99% being rare, the ability to detect differences in expression of mRNA at low levels is crucial.
DIFFERENTIAL DISPLAY
Differential display PCR and its related technologies involve the RT-PCR amplification of two different mRNA populations using defined oligomers followed by separation of the resulting fragments side by side on a denaturing polyacrylamide gel.9 ,10Differentially expressed bands at different points on the gel are removed and re-expanded by PCR and cloned. A number of variations of this technology have been developed to try to overcome the main problems—namely, the labourious nature of the approach, the generation of false positive bands, and the limited ability to quantify levels of mRNA expression. It is available in kit form.
REPRESENTATIONAL DIFFERENCE ANALYSIS (RDA)
RDA is an alternative method which is aimed at reducing the number of candidates genes. It combines a subtractive hybridisation step with amplification using PCR, thus optimising for differences.11-13 Again, a number of variations have been developed.
SERIAL ANALYSIS OF GENE EXPRESSION (SAGE)
SAGE was developed by Kinzler, Vogelstein and Velculescu and involves the isolation of unique 3′ sequence tags which are then concatenated and sequenced.14 ,15 Thus, each individual clone that is sequenced contains many partial length tags (about 10–50). Importantly, the frequency of many transcripts and the starting mRNA pool can be determined by the frequency with which a specific sequence tag is found in the sequenced population. SAGE has advantages in terms of efficiency, quantification, the ability to detect novel genes and, importantly, the ability to detect low abundance transcripts (table 1).
Advantages and limitations of SAGE and DNA chip technology for analysis of difference in mRNA expression in different tissues
The success of SAGE largely relies on the availability of sequences representative of genes in the particular target species. Such genes are largely in the form of expressed sequence tags (ESTs). There are now a large number of ESTs in public and private databases. Approximately 60% of human genes are now currently available while a lesser proportion of murine genes are available on database. There are also some large public and private “warehouses” containing large scale genetics databases—for example, Incyte Genetics.
The application of this technology to two different pulmonary tissues/samples/conditions may run the risk of producing large numbers of sequence differences without there being a readily available database to analyse the significance of these differences. Thus, it is possible to generate a large amount of information that is, at this stage, difficult to interpret.
SAGE has some advantages over DNA chip arrays in that novel genes can be discovered and more sequences can be analysed. SAGE can be used in multiple experiments. Genzyme has acquired the commercial rights to SAGE technology. SAGE analysis of lung cancer has recently been published.16
SEQUENCING OF EXPRESSED SEQUENCE TAGS (EST)
Single pass sequencing of the 5′ or 3′ ends of cDNAs from libraries representing various tissue and cell types has been used to define sequence tags that uniquely identify a particular gene and tags in the public domain are currently available through dbEST.17-20 The enumeration of the relative representation of these unique tags within a cDNA library derived from a particular tissue or cell type has been used as a method of approximating the relative representation of the gene transcript within the starting cell population. SAGE is more efficient than EST sequencing when comparable sequencing approaches are compared.
Microarray analysis (“DNA chip technology”)
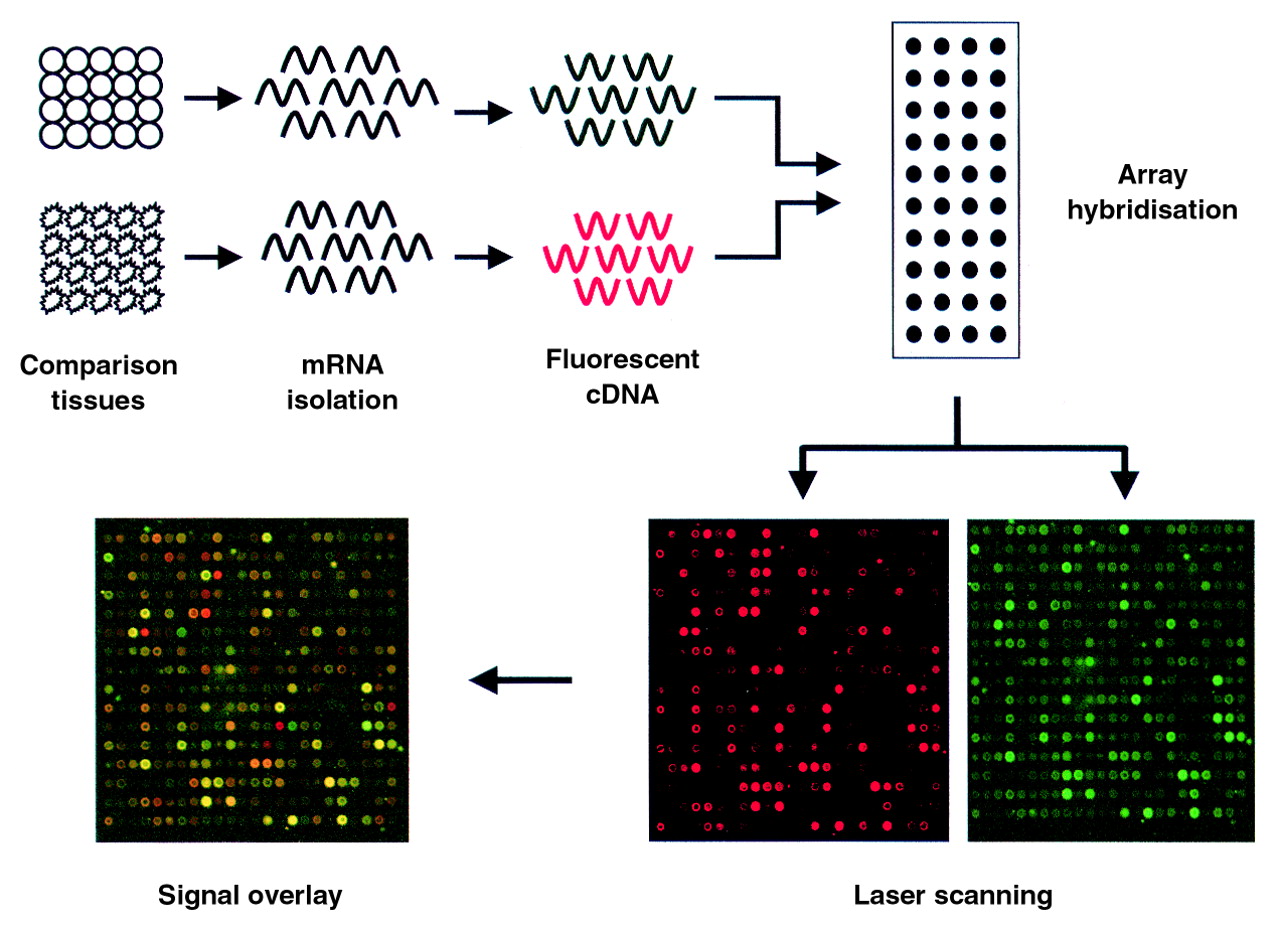
In microarray expression analysis DNA chip technologies allow the parallel analysis of thousands of mRNAs and differ from the above techniques in that they provide expression measurements on defined sets of genes.21 ,22 Chip techniques generally involve the arraying and deposition of target cDNAs or oligonucleotides onto support matrices that include nylon filters, microscope slides, or silicon chips.21 ,22 Differential mRNA analysis using DNA arrays is accomplished by hybridising the target array against total cDNA pools derived from particular cell or tissue types of interest. The quantity of probe bound to each target cDNA on the array is measured using radiolabelled tracers or fluorescent tags and reflects the relative abundance of its corresponding message in the cells or tissues from which it was derived. The overall hybridisation to the array provides a profile of the relative message levels for each gene represented on the microarray (fig 3). Difference analysis is accomplished by comparing the hybridisation signals generated by each mRNA/cDNA sample at each spot on the array—for example, signal derived from normal versus diseased tissues.23-30 Since high density microarrays can currently contain up to 10 000 distinct genes, a large portion of the expressed genome can be compared in a single experiment. It is hoped that this technology will help investigators to identify therapeutic targets,24 ,29 develop new approaches for diagnosing disease,24 and understand the pharmacological and toxicological impact of new drugs on human tissues.31-33
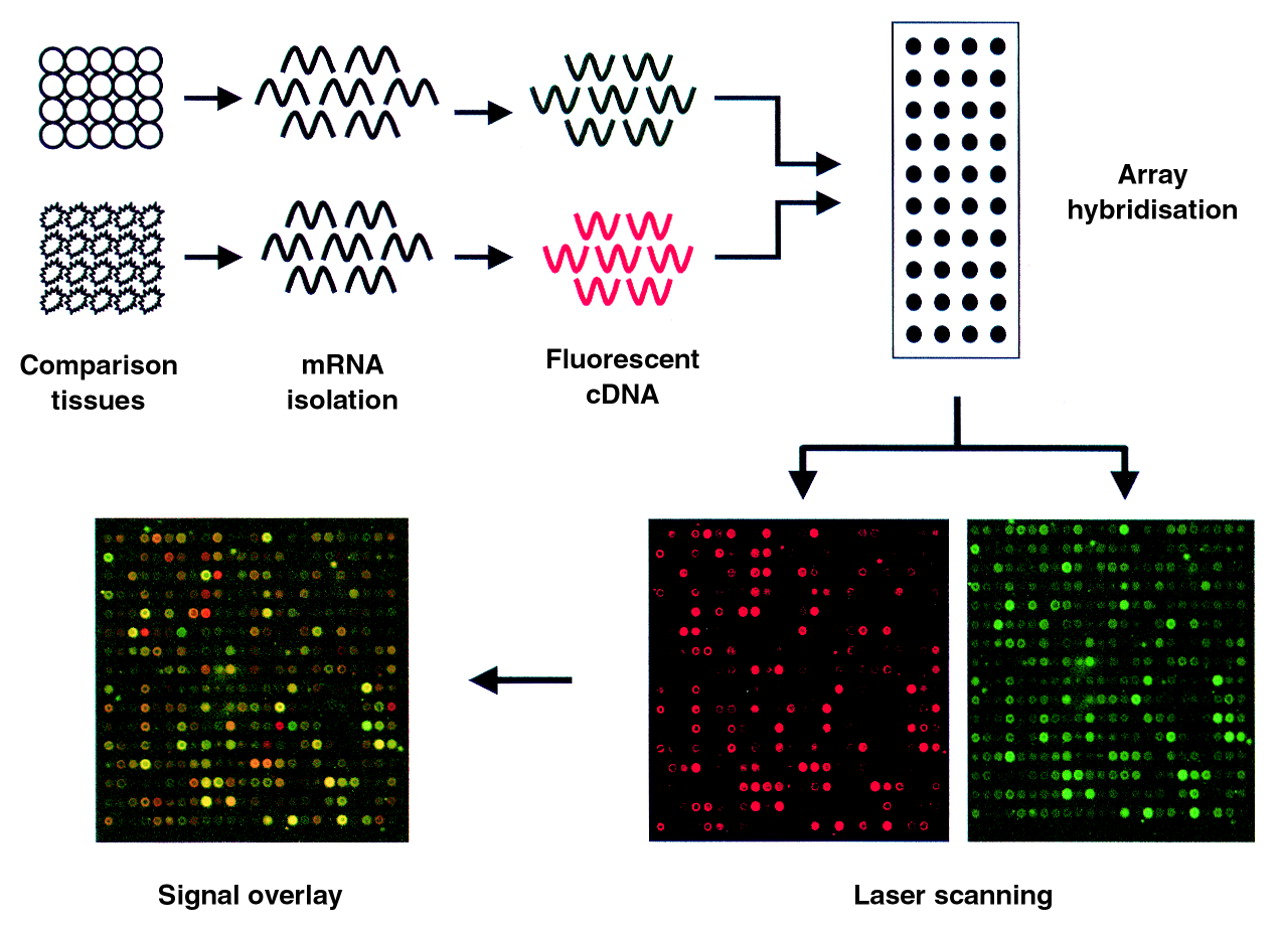
Schematic representation of microarray profiling. mRNAs from two sources are labelled with distinct fluorescent tags by reverse transcription and mixed in equimolar amounts. The mixture is then hybridised to a slide spotted with distinct cDNAs. Following hybridisation, laser scanning on channels specific for each tag quantifies the hybridisation intensities at each spot for each probe pool. The resulting hybridisation pattern reveals an “activity signature” for the subject comparison. Ratios of the two signals can be calculated and represented graphically to determine gene expression differences.
DNA microarrays were used recently by Karpf et al to examine the response of human tumour cells to the chemotherapeutic agent 5′-aza-2′-deoxycytidine.33 This compound is under investigation as a potential therapeutic agent in lung and prostate cancers and may elicit beneficial and toxic effects by inducing the expression of genes silenced by promoter methylation.34 A comparison of the transcript profiles of colon carcinoma cells treated with 5′-aza-2′-deoxycytidine and control untreated cells revealed a distinctive pattern of gene expression that suggested activation of interferon signalling pathways. Follow up analyses revealed that treatment of the cells with 5′-aza-2′-deoxycytidine stimulated the expression and activation of the transcription factors, STATs 1, 2 and 3. This suggested STAT expression levels as rate limiting determinants of interferon responsiveness and raises the possibility that treatment of tumours with 5′-aza-2′-deoxycytidine increases their sensitivity to interferons. These observations offer two important points regarding the use of microarrays for evaluating compounds and targets. Firstly, they show that microarray data can generate testable molecular hypotheses regarding disease development and, secondly, they illustrate how transcriptional responses may reflect and explain compound efficacy and toxicities. Analyses on DNA chips are currently limited by the number of discrete known genes available for array and are yet to be developed to the point where large numbers of such chips are available commercially. Furthermore, the development of gene sets for species other than human, yeast and C elegans are lagging. A number of products are now available commercially for performing array experiments including small arrays covering specific gene/functional families (apoptosis, cell cycle, adhesion) and larger randomly selected gene sets arrayed on both chips and nylon membranes. The main limitation that exists in terms of applying these technologies to pulmonary disease, particularly tissue biopsy, is the relatively large amounts of mRNA required for array hybridisation. However, as cDNA library sources grow, specialised, disease specific arrays requiring smaller sample amounts are likely to alleviate this limitation. Robotic chip “spotters” for glass slides and chip scanners for array analysis are available commercially.
Overall, it is hoped that this technology will help investigators to identify therapeutic targets, develop new approaches for diagnosing disease, and understand the pharmacological and toxicological impact of new drugs on human tissues. These databases are, of course, limited to known genes and are yet to be developed to the point where a large number of such chips are available which represent different species or different groups of clones.
With further advances in this technology, which will occur quickly, it will be possible to generate huge amounts of information on differences between separate biological samples. Other applications include the labourious task of polymorphism discovery and its correlation with diseases and drug effects. It is hoped that polymorphism analysis will enable investigators to differentiate individuals according to the likelihood of them suffering diseases or side effects of treatment.35 ,36 As mentioned above, however, the analysis of these data will be more critical than its generation. As a consequence of this, and as a byproduct of the human genome project, the use of “bioinformatics” has developed to facilitate the analysis of data generated using such technology.37
Scientists with experience in the analysis of pathological processes who have access to high quality representative samples of the disease and relevant control tissues will be in a strong position to utilise the information generated by these approaches. The general term being used to describe the process of understanding the roles of the genes and products detected using new molecular technologies is “functional genomics”.38
APPROACHES TO TISSUE ANALYSIS
It is important to mention two broad philosophical approaches to the use of these types of technologies. The first involves a largely “reductionist” approach in which biological factors that generate complexity in a given tissue reaction are removed from the process, providing a simple analysis of responses. The second approach involves the analysis of complexity, either in a model system or usually in vivo, in which most of the interacting biological factors are present. In pulmonary disease this has involved analysing animal models, human lung tissue, bronchoalveolar lavage (BAL) fluid samples, etc. The complexity of the system makes such analysis difficult, but the general aim of this approach is to fulfill a modified version of Koch's postulates—that is, to attempt to define the role of a factor in pulmonary disease by showing that it is present at the time of the disease, that its removal prevents or reduces the disease (for example, antibodies, antisense, knockout mice, etc), and that, when added back to the system, the disease process is reproduced.
TARGET PULMONARY TISSUES
Human tissue
Human lung tissue is extremely complex and it is difficult to obtain pure, non-cultured cell preparations from specific lung components. Laser dissection is one method of doing this. It is essential that any tissues being analysed in these new technologies are very fresh in order to preserve mRNA; however, high quality human lung tissue can be difficult to obtain. Samples from resected specimens are often not fresh, even if obtained in the operating theatre. In any case, pathologists like to have “first go” at the tissue and, understandably, do not like to see important pieces of the sample missing before they begin their analysis. This applies particularly to tumour resection samples. Of course, such samples also have the limitation created by the presence of the underlying disease—for example, smoking-induced changes in patients undergoing resection for lung malignancy. Biopsy specimens such as bronchial, pleural, or transbronchial lung tissues can be useful, particularly if the diagnosis has already been established, as the whole sample can be used and these samples are more readily preserved in OCT, RNAzol, etc. BAL has been a useful tool for the past 20 years or so39 and has the advantage that cells are obtained fresh and already separated and, at least in diseases such as sarcoidosis, seem to be reasonably representative of the cells in the tissue. mRNA in these cells, however, is rapidly degraded. Similarly, pleural fluid represents a good source of already separated cells. Sterile samples that are anticoagulated can provide large numbers of tumour and inflammatory cells, although there has been no detailed functional analysis of effector cells to determine whether or not they are representative of those present in pleural tissue. Blood samples are of limited value because the mononuclear cell populations do not usually reflect those present in the tissue, and because most immunological relevant molecules such as cytokines are only released in high concentration in the tissue of origin and are either not found in the circulation or are bound or degraded in the blood.
Animal tissue
Murine and other animal models of pulmonary disease have been widely utilised. Although they are never perfect models of the corresponding human pulmonary disease, there are a number of models in which the clinical behaviour, histopathology, immunology, and cell biology accurately parallel human disease. Thus, they provide useful information in diseases such as asbestos induced parenchymal disease and pleural tumours. Importantly, animal models provide the opportunity of fulfilling the modified Koch's postulates described above—that is, they are able to remove or add back various factors in the animal model using monoclonal antibody blockade, anti-sense technologies, gene transfer, and transgenic/gene knock out approaches.
“Contaminating cells” remain one of the biggest drawbacks of tissue analysis from human or murine tissue. A small proportion of contaminating cells in a given tissue could provide a huge number of different mRNAs which will not add substantially to the data—for example, contaminating bacteria or different cell profiles in BAL fluid samples will generate a lot of “difference” which may not be relevant to the disease process.
Proteomics
Proteomics is less well developed. Its principle is similar to the difference analysis described above except proteins, rather than mRNAs, are targeted. Interestingly, the most abundant mRNAs are secreted proteins whereas the most abundant proteins are not secreted. Proteomics aims to study differential protein expression and protein interactions. As about 10–30% of genes are expressed in cells, at least 104 proteins are produced, providing the opportunity for at least 1016 protein–protein interactions between just two expressed products. Proteomics relies heavily on 2D gel electrophoresis and careful analysis of the different spots obtained when comparing two different samples.40 ,41 Of course, it has the same difficulties that are described above for mRNAs—that is, each individual cell generates a huge number of different proteins and it is difficult to get clear results in samples other than with microorganisms. As with difference analysis, proteomics data rely on sophisticated bioinformatics to generate models and to recognise a “cluster” expression pattern.
Transgenic and gene targeted mice
Within the next few years the sequence of all human genes will be known, but the function of proteins encoded by most of these genes will remain a mystery. Gain of function (transgenics) and loss of function (gene targeting) models provide powerful means to perform controlled experiments in mammals in vivo and hence determine protein function. Techniques to achieve germline transmission of genetic material in mice have drastically changed our ability to approach biological questions. Introduction of a linear DNA fragment (transgene) into the pronucleus of one celled embryos (or, more recently, into embryonic stem cells) allows study of either the pattern of expression of that gene or the biological consequences of overexpression of the protein encoded by the gene in specific tissues. More recently gene targeting or targeted mutagenesis by homologous recombination in embryonic stem cells has allowed investigators to generate strains of mice that lack individual proteins, providing specific loss of function models.
Mice are used mainly because of the unique capacity to achieve germline transmission of genetic information; however, additional advantages of the mouse over other experimental animals include a rapid reproductive cycle and large litter sizes. In addition, knowledge of mouse biology is good, antibody and cDNA probes for the mouse are abundant, and mice are relatively cheap compared with other experimental animals. Importantly, evolutionary conservation has shown that mice and other mammals are embarrassingly similar to humans. On the other hand, the applicability of these studies to understanding human biology and the dissect mechanism of disease requires knowledge of similarities and differences with respect to the protein profile between mouse and humans. Also, murine models should closely replicate the human disease.
Determining which proteinases may contribute to pulmonary emphysema provides an example of these concepts. Pulmonary emphysema is defined as enlargement of terminal air spaces accompanied by destruction of their walls. Emphysema is a major component of the morbidity and mortality of chronic obstructive pulmonary disease (COPD) which is currently the fourth leading cause of death in the USA and one of the causes whose incidence is increasing. COPD may grow to epidemic proportions given the large increase in smoking worldwide.
Typically, cigarette smoke induces a chronic inflammatory response within the lung. Release of inflammatory cell proteinases, in excess of inhibitors, coupled with abnormal matrix repair leads to emphysema. To determine the contribution of individual proteinases we found that exposure of mice to cigarette smoke results in inflammatory cell recruitment and air space enlargement similar to humans. We have now applied several proteinase deficient mice to this model. The most striking phenotype was observed in mice lacking macrophage elastase (MMP-12).42 Mice deficient in macrophage elastase (MMP-12–/–) were entirely protected from development of emphysema despite heavy long term smoke exposure. Surprisingly, MMP-12–/– mice also failed to recruit monocytes into their lungs in response to cigarette smoke. Our working hypothesis is that cigarette smoke induces constitutive macrophages, which are present in lungs of MMP-12–/– mice, to produce MMP-12 that in turn cleaves elastin thereby generating fragments chemotactic for monocytes. This positive feedback loop perpetuates macrophage accumulation and lung destruction. The concept that proteolytically generated elastin fragments mediate monocyte chemotaxis is not original. Independent studies by Senior and colleagues43and by Hunninghake et al 44 from the early 1980s demonstrated that elastase generated elastin fragments were chemotactic for monocytes and fibroblasts. Gene targeting is merely reinforcing this as a major in vivo mechanism of macrophage accumulation in a chronic inflammatory condition. Whether human emphysema is also dependent on this single MMP is, of course, uncertain. At the very least, this study demonstrates a critical role of macrophages in the development of emphysema and unmasks a proteinase dependent mechanism of inflammatory cell recruitment that may have broader biological implications.
Techniques are rapidly being developed to make mutations specifically when and where desired (fig 4).45 Use of tetracycline to achieve inducible overexpression remains difficult, but successes are powerful. The tool that appears to be most powerful for conditional gene deletion is the bacteriophage Cre recombinase, which lines up and removes sequences bracketed by Lox P sites.46 This technology can be used in ES cells to remove PGK-neomycin selectable marker (which interferes with gene expression), allowing other genes to be “knocked in” to the locus of another gene. One can also use Cre-Lox technology to perform tissue specific knockouts by crossing transgenic mice expressing Cre recombinase in a tissue specific fashion with mice that have LoxP sites located in introns flanking coding sequences. Only those tissues expressing Cre recombinase will delete the gene of interest. Strategies are also underway using Cre linked to steroid receptor proteins to achieve inducible and tissue specific gene targeting. In summary, methods are rapidly being developed to overexpress and delete genes under both spatial and temporal control. Unlimited possibilities will soon be practical, limited only by the investigator's imagination, time, and resources.
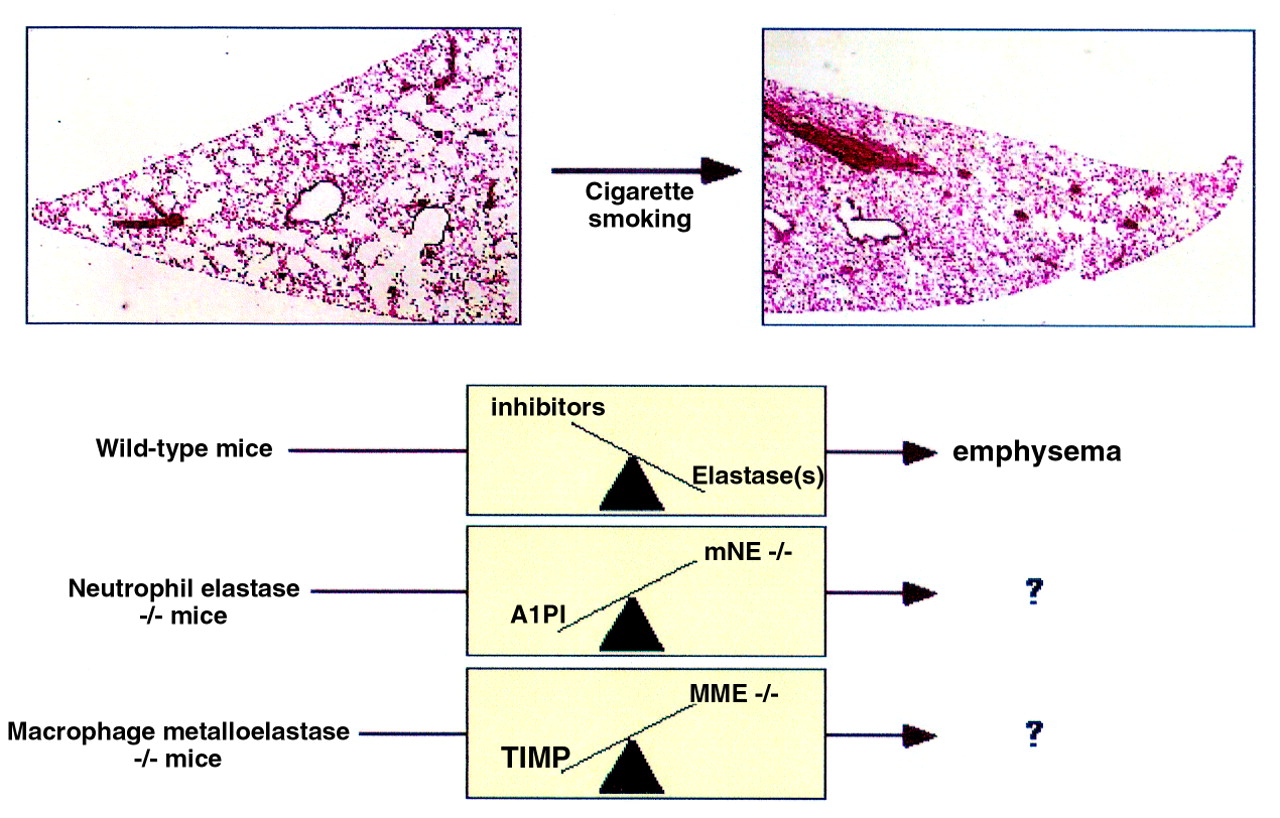
Murine model of cigarette induced emphysema and application to elastase deficient mice. Low power magnification (×40) of haematoxylin and eosin stained lung sections obtained from a wild type C57BL/6 (non-smoking ) control mouse (left) and a mouse exposed to cigarette smoke for six months (right). Note enlarged lung volumes (both inflated 25 cm H2O) and enlarged alveolar spaces characteristic of emphysema in smoke exposed mouse. The elastase:antielastase hypothesis would predict that lung destruction in wild type mice was secondary to a shift in the balance favouring unopposed elastase activity (depicted by scale). Application of this model to mice deficient in individual elastases including mouse neutrophil elastase (mNE–/–) and macrophage elastase (MME–/–) has uncovered roles for both of these proteinases, particularly MME in emphysema. Studies also predict much more complicated scenarios than the simple elastase:antielastase balance depicted here.
Antisense technology
A number of new molecular tools have recently been developed aimed at blocking the function of specific molecules in vivo. Some of these include humanised monoclonal antibodies, small molecular receptor antagonists, and inhibitors of cellular adhesion. This section covers another area—epigenetic attenuation of signals in lung diseases—using allergic disease as a model. This form of modification of genetic translation by reducing protein products uses antisense oligonucleotides (ASONs) (fig 5). These are short oligonucleotides, modified to slow degradation, which code for peptides that match the non-coding strand of DNA. When introduced into the cell they hybridise with messenger RNA to prevent the production of the protein through a “translational arrest”. Advantages of antisense technology include its high specificity, its ability to be delivered locally to the lung (for example, respirable antisense oligonucleotides (RASONs)), reduced cost of production, and lack of systemic side effects. Several disadvantages include tedious assays to assess the specificity and effectiveness of the antisense, finding the appropriate antisense on a three-dimensional RNA structure, and the potential generation of DNA antibodies.47 The primary advantage is the specificity compared with other oligonucleotide therapies. These use the CPG motif48 found in bacterial DNA to enhance “molecular immune surveillance”49 ,50 and modify the imbalance between TH1 and TH2 subsets. Asthma is unique because it allows the administration of antisense directly into the lung through a nebuliser or metered dose inhaler where surfactant51 may enhance its uptake.

Antisense oligonucleotides (ASONs). ASONs are incorporated intracellularly, hybridise with mRNA and, through degradation by ribonuclease H, cause translational arrest and subsequent attenuation of receptor protein. Adapted with permission from Metzger and Nyce.50
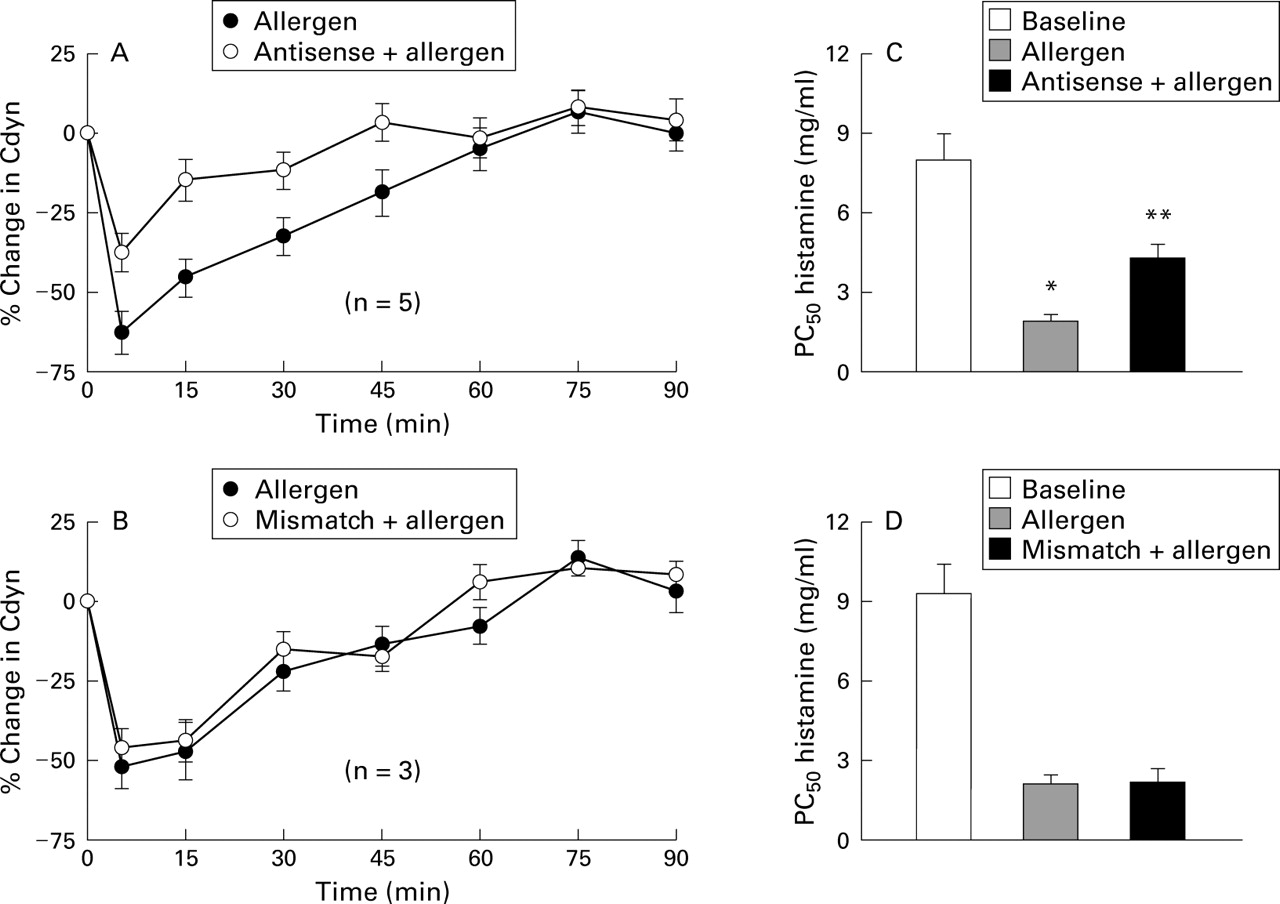
A human antisense against the adenosine A1 receptor (EPI-2010) reported by Nyce and Metzger52 was recently tested in an allergic rabbit model for early and late phase allergic airway inflammation. Adenosine A1 receptors have been shown to be upregulated in this rabbit model.53 The serendipitous homology between the human A1 receptor and rabbit A1 receptor made the allergic rabbit an excellent testing ground for this new technique for modulating lung receptors. Initial studies administered a total of 20 mg A1 antisense EPI-2010 (10 mg each day in divided doses) to rabbits allergic to house dust mites in a randomised crossover protocol.52 In these studies adenosine A1 receptor binding activity and A1 receptor number were reduced significantly (75%) by administration of antisense by direct inhalation of the nebulised material. A1 antisense showed no effect against the A2 receptor and bradykinin B2 receptor. Mismatch controls for the A1 receptor and bradykinin B2 receptor showed no effect on receptor binding or downregulation of the A1 receptor number. The RASON EPI-2010 also reduced bronchial hyperresponsiveness and immediate airway obstruction in rabbits sensitised to house dust mite (fig6).52 More recent studies have shown treatment with aerosolised EPI-2010 to be effective in reducing adenosine induced bronchoconstriction in the allergic rabbit for an average of 6.8 days, with some animals having a modified response for up to 10 days.54 Further studies using radiolabelled EPI-2010 showed that the RASON was effectively confined to the lung without appreciable deposition in any other tissues.55 Deposition in the lung was uniform and found in both peripheral and central airways. The half life for EPI-2010 was approximately 30 hours with elimination measured primarily through the urine.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of EPI-2010 ASONs in vivo. ASONs and mismatch control ODNs (ODNs differing by one or two sequential base pairs) were evaluated in allergen induced airway obstruction and bronchial hyperresponsiveness (BHR) in allergic rabbits. (A) Effect of A1 antisense ODN on allergen induced airway obstruction as measured by change in dynamic compliance (Cdyn). As calculated from the area under the curve, EPI-2010 antisense significantly inhibited allergen induced airway obstruction (55%, p<0.05; repeated measures ANOVA and Tukey's t test); (B) lack of effect of mismatch control A1 ODN on allergen induced airway obstruction; (C) effect of EPI-2010 antisense on allergen induced BHR. As calculated from the PC50 for histamine, EPI-2010 antisense significantly inhibited allergen induced BHR in allergic rabbits (61%, p<0.05; repeated measures ANOVA, Tukey's t test); (D) lack of effect of mismatch A1 ODN control on allergen induced BHR. Reprinted with permission from Nyce and Metzger.52
These data suggest that antisense for the A1 receptor may be useful in the treatment of asthma. In addition, single dosing once a week may become the standard for RASONs. The small doses administered to the lungs should have little or no toxicity systemically, which has been a problem with systemic administration of antisense for other applications such as HIV, cancer and organ transplantation, and neurological diseases.56 The potential for RASONs in respiratory disease is extremely broad and includes lung cancer, COPD, and bronchial hyperresponsiveness in addition to asthma. It may even be extended to include rhinosinusitis. The range of antisense development is seemingly unending, but antisense against such key players as IL-4, IL-5 or CCR3 chemokine receptor may be uniquely useful to modify the IgE mechanism of allergic disease. RASONs therefore represent great potential for the treatment of allergic diseases with enhanced efficacy, specificity, and safety. The proof of this awaits human studies, which are currently in progress.
Gene delivery strategies in mammalian cells
The expression of proteins in mammalian cells from cloned DNA is one of the most powerful techniques in molecular biology. Depending on the goals of the experiment, different approaches can be used (table2). In in vitro experiments the most important distinction is between transient transfection and the creation of stable cell lines expressing the gene of interest. The methodology for performing gene transfer experiments is now standard and well described in one of many textbooks of molecular biology techniques.57
Comparison of in vitro gene transduction techniques
In transient transfection cDNAs are expressed at high levels but only for a short period of time (days) in a subpopulation of cells. Plasmids can be introduced into the cells using calcium phosphate precipitation, DEAE-dextran, or lipofection. The advantages of this approach are that it is quick and reliable (transfection and analysis usually completed in one week), cell lines commonly used are very easy to transfect, large amounts of protein can be produced, and vectors are easy to subclone into and use. On the other hand, no stable cell lines are produced (so the whole transfection must be repeated if new cells are needed), only subpopulations of cells (10–30%) express the desired protein, the number of cell types that can be used for these experiments are limited, and post-translation protein processing may be different from that in the “normal” cell host. Transient transfection is commonly used to confirm the identity of a cloned gene using immunological or functional assays to detect the encoded protein, to perform “expression cloning” where cDNAs can be isolated by functional criteria,58 to produce large amounts of proteins of biological interest, to study structure/function relationships of proteins by analysing the properties of normal and mutant proteins, to produce mutant proteins that allow “epitope” mapping of bioactive monoclonal antibodies, and to identify DNA sequence elements involved in control of gene expression.
In permanent transfection proteins are permanently expressed at lower levels but (after selection) in a very high percentage of the cells. Cell lines are commonly permanently transfected using calcium phosphate precipitation, DEAE-dextran, lipofection, or electroporation. The plasmid must contain the gene of interest along with a selection marker or it must be transfected along with a second plasmid containing the selection marker. One increasingly important variant of this approach is to use retroviral vectors into which is cloned the gene of interest. After transfection of cells with a retrovirus containing the transgene and a selection marker, the cells are selected in much the same way as described above. One major advantage is that retroviral transduction is often much more efficient than any of the other techniques due to receptor mediated uptake. Advantages of permanent transfection are that cell lines are produced that often stably express the gene of interest at high levels indefinitely; so cells can be frozen and stored. This stability is critical in studies where cells are introduced into animals. These cell lines can be used for functional assays where it is important for a large percentage of cells to express protein and cell lines with varying levels of expression can be isolated. A much wider variety of cells can be transfected and studies on protein processing can be performed. Disadvantages of this approach are that it is time consuming, more technically challenging (often 2–3 months between transfection and isolation of highly expressing clone), and some cell lines are quite resistant to transfection and/or express transgene at low levels. Cells to be transfected must have sufficient “lifespan” in culture to allow selection from clonal density.
Adenovirus transfection is a newer and somewhat hybrid approach. Genes can be expressed at high levels with very high efficiency in a large population of cells for periods of time ranging from two days to two weeks. Adenoviral vectors that are rendered replication defective are made by homologous recombination in 293 cells, plaque purified, and grown up in large quantities.59-62 This can be done using a plasmid system or viral DNA. Commercial systems are now available. Transfection is simple and, with titration of the virus, virtually 100% of most cell lines can be transfected. Adenoviral transduction is quick and reliable (transfection and analysis usually completed in one week), no selection is needed, most cell lines commonly used are relatively easy to transfect (including a number of non-transformed cell lines such as endothelial cells that are very difficult to transfect otherwise), large amounts of protein can be produced, and high levels of protein expression can be obtained in most cells for up to two weeks. The disadvantages are that no stable cell lines are produced, a heterogeneous mixture of cells results from transduction, preparation of vectors is technically difficult and time consuming (1–3 months), some cell lines are resistant to adenoviral transfection, and viral proteins can be toxic to some cells.
In addition to transfection or viral introduction of DNA expression vectors, it is also possible to introduce proteins directly into cells (protein transfection). Until recently this was only possible by microinjection techniques, a time consuming and tedious approach that only allowed small numbers of cells to be “transfected”. Over the past 10 years, however, specialised domains have been identified in the HIV tat protein and in the Antennapedia (Antp) protein ofDrosophila that allow intercellular transport. Accordingly, fusion proteins using these key sequences can be introduced into cells.59 Another protein with intercellular transport properties is the herpes simplex virus protein VP22.63 When cells were transfected with cDNA encoding fusion proteins of VP22 with green fluorescent protein (GFP), many cells adjacent to transfected cells also expressed. This approach has appeal to gene therapy where “bystander” effects are very advantageous. A plasmid containing VP22 followed by a cloning site is now commercially available from Invitrogen.
Many of these same techniques are being used for in vivo transfection (gene therapy) to the lung for diseases such as cystic fibrosis, lung cancer, and malignant mesothelioma.64 To date, the most effective strategies have been liposomal gene transfer and the use of adeno associated viruses and adenoviruses.65 Although beyond the scope of this review, table 3 (adapted from Curielet al 64) summarises some of the most important characteristics of commonly used gene therapy vectors. The major problems continue to be difficult to solve—namely, delivery (getting the transgene into a large proportion of the target cells but not other cells), efficiency (efficient production of a large amount of relevant mRNA in the target cells with the production of large amounts of the target protein), and durability (avoiding the many worries of switching off transgene expression, particularly with the generation immune responses). These are areas of active research and will be critical if gene therapy for the lung is to be successful.
Characteristics of gene therapy vectors (adapted from Curiel et al64)
Conclusions
The advances described have the capacity to provide a large amount of important new information relevant to pulmonary disorders. Once established in a laboratory, it will be crucial to select the relevant tissues and to have in place valid ways of identifying important differences to avoid the accumulation of uninterpretable masses of data. Because these technologies can provide accurate high analytical capabilities, it is possible that research can shift from “hypothesis driven research” to “technology driven research”, in the process generating masses of confusing data. It is essential that the strengths and weaknesses of these new technologies are understood to avoid the generation of large amounts of largely uninterpretable data. This will involve careful selection of target pulmonary tissue and control tissue, detailed attention to the handling of that tissue, selection of techniques which can provide accurate and reproducible information and, importantly, access to sophisticated bioinformatics technology that will enable the data to be meaningfully analysed.
Glossary
- Antisense oligonucleotides:
- short oligonucleotides (8–21 mer) which code for peptides that match the non-coding strand of DNA; when introduced into a cell they hybridise with mRNA to prevent production of the protein through “translational arrest”.
- Bioinformatics:
- computer analysis of biological information.
- Competitor PCR:
- PCR involving the amplification of two different DNA species, usually of different sizes, that both contain sequences recognised by the primers. Used for quantitative PCR.
- DNA chip technology:
- the arrangement of cDNAs or oligonucleotides on silcone chips for comparative tissue analysis.
- Epigene therapy:
- modification of genetic machinery to modulate the results of translation into products.
- EST:
- expressed sequence tags; 3′ or 5′ ends of cDNAs representing various tissue types.
- Functional genomics:
- analysis of the role of genes and gene products.
- Homology based PCR:
- PCR performed using primers designed based on conserved regions of homologous proteins.
- PCR (polymerase chain reaction):
- performed on RNA or DNA; a “molecular photocopier”.
- Permanent transfection:
- expression of cDNAs expressed stably after selection in a very high percentage of the cells.
- RASONs:
- respirable antisense oligonucleotides.
- RDA:
- representational difference analysis; combines a subtractive hybridisation step with PCR amplification.
- SAGE:
- serial analysis of gene expression. A technique using concatenation of small cDNA expressed sequence tags; a quantifiable method of difference analysis.
- Transient transfection:
- expression of cDNAs at high levels but only for a short period of time (days) in a subpopulation of cells.
Some relevant web sites
pcr http://www.perkin-elmer.com http://www.thoracic.org/assemblies/rcmb/erle.htm
sage http://www.genzyme.com/prodserv/molecular_oncology/sage/welcome.htm http://www.sagenet.org/
dna chips http://www.ncbi.nlm.nih.gov/ncicgap/EST/cgapqr.cgi http://www.ncbi.nlm.nih.gov/ncicgap/expression_tech_info.html http://www.synteni.com/ http://www.incyte.com/ http://www.hyseq.com/ http://www.affymetrix.com/
antisense http://www.epigene.com http://www.oligosetc.com http://www.genosys.com
gene therapy http://www.asgt.org/ http://www.wiley.co.uk/genetherapy/ http://www.uiowa.edu/∼gene/