Article Text

Statistics from Altmetric.com
Systemic lupus erythematosus (SLE), an autoimmune disorder which primarily affects women (10:1 female to male ratio), may affect virtually any organ.1 Predominant manifestations include non-deforming arthritis, serositis, photosensitivity, renal, haematological, and central nervous system involvement. Various laboratory abnormalities have been described in SLE, most commonly high titre antibodies directed against double stranded DNA,1nuclear ribonucleoprotein, Smith (Sm) antigen, Ro/SS-A, and La/SS-B/Ha.2 Pleuritis occurs in 17–60% of patients at some point in the course of the disease.1 ,3 Although severe parenchymal lung disease is uncommon, pulmonary complications of SLE are protean and include acute lupus pneumonitis, diaphragmatic dysfunction and shrinking lung syndrome, cavitating pulmonary nodules, pulmonary hypertension, pulmonary vasculitis, pulmonary embolism (often due to circulating anticardiolipin antibodies), alveolar haemorrhage (reflecting diffuse endothelial injury), chronic interstitial pneumonitis,3-7 bronchiolitis obliterans (with or without organising pneumonia),8 and opportunistic pulmonary infections or drug toxicity from immunosuppressive therapy.9
Pleural disease
The most common thoracic manifestation of SLE is pleuritis.3 ,10 Pleuritic pain is present in 45–60% of patients and may occur with or without a pleural effusion (fig1).3 Clinically apparent pleural effusions have been reported in up to 50% of patients with SLE3 and may be found in up to 93% of cases at necropsy.11 Effusions are usually bilateral but may be unilateral, equally distributed between the left and right hemithoraces. They are invariably exudative with higher glucose and lower lactate dehydrogenase levels than those found in rheumatoid arthritis.12 Antinuclear antibody (ANA), anti-DNA antibodies, and LE cells have been found in the pleural fluid.13 ,14 LE cells appear to be relatively specific.15 Pleural biopsies have rarely been performed in SLE and the findings are non-specific, revealing lymphocytic and plasma cell infiltration, fibrosis, and fibrinous pleuritis.16They are indicated primarily to exclude alternative aetiologies. Thoracoscopy has revealed nodules on the visceral pleura and immunofluorescence of biopsy samples of these nodules revealed immunoglobulin deposits.17

Chest radiograph showing left pleural effusion in a 50 year old man with arthritis, fever, and high titre anti-DNA. Thoracentesis demonstrated typical LE cells. Reproduced with permission from Orens et al.3
Treatment of pleural disease depends on the severity of symptoms. Small asymptomatic effusions may not require specific treatment. Non-steroidal anti-inflammatory agents may be effective for mildly symptomatic pleurisy. For more severe disease or in patients already receiving corticosteroids, increasing doses of corticosteroids are frequently required.3 ,18 Long term management may involve the use of antimalarial drugs such as hydroxychloroquine. Invasive procedures such as chest tube drainage or pleurodesis are rarely required.19
Chronic interstitial pneumonitis (CIP)
Clinically significant chronic interstitial pneumonitis (CIP) complicates SLE in 3–13% of patients but is rarely severe.1 ,3-6 Asymptomatic involvement is more common and abnormalities in pulmonary function tests have been cited in up to two thirds of patients with SLE in some studies.3 ,20Progression of recurrent acute lupus pneumonitis to CIP probably occurs since there are examples of persistent disease following acute onset, and the mean age of onset of acute disease (38 years) is earlier than that for chronic disease (46 years).2 Chest radiographic abnormalities consistent with CIP have been cited in 6–24% of unselected patients with SLE.20 ,21 A recent prospective study assessed chest radiographs, pulmonary function tests, and high resolution computed tomographic (HRCT) scans in 34 patients with SLE.20 Abnormalities in HRCT scans or chest radiographs were detected in 24 and eight patients, respectively.20Eleven patients were judged to have interstitial lung disease on HRCT scanning, nine of whom were asymptomatic. Pulmonary function tests were abnormal in seven of the 11 patients. Subclinical CIP may be common, diagnosis depending on the sensitivity of the diagnostic tests used, and the long term significance of this has not been elucidated. However, symptomatic CIP is rarely an early or dominant feature of SLE and severe pulmonary fibrosis is rare (fig2).1 ,3 Moderate or severe pulmonary fibrosis was present in only four of 120 necropsy specimens in patients with SLE.4 Histological features of CIP complicating SLE are non-specific and include varying degrees of chronic inflammatory cell infiltrates, peribronchial lymphoid hyperplasia, interstitial fibrosis, and hyperplasia of type II pneumocytes.3Clinical and serological parameters fail to correlate with the nature or extent of pulmonary functional abnormalities.3 An association between anti-SS-A antibodies and chronic lupus pneumonitis was suggested in one study in which 81% of patients with lupus pneumonitis had anti-SS-A (Ro) antibodies5; others found no correlation between serological abnormalities (anti-SS-A, anti-SS-B, anti-Sm) and pulmonary function tests.22 Anti-U1 RNP antibodies were associated with reduced lung volumes and transfer factor in one study; Sm antibodies did not correlate with pulmonary functional impairment.23 The presence of scleroderma-like traits—for example, Raynaud's phenomenon, swollen fingers, sclerodactyly, telangiectasia, dyspnoea, nailfold capillary abnormalities—among patients with SLE was associated with a higher prevalence of restrictive defects or reduced transfer factor.23 Progressive severe CIP rarely complicates SLE but may be seen in a subset of patients with SLE in the context of overlap syndrome.
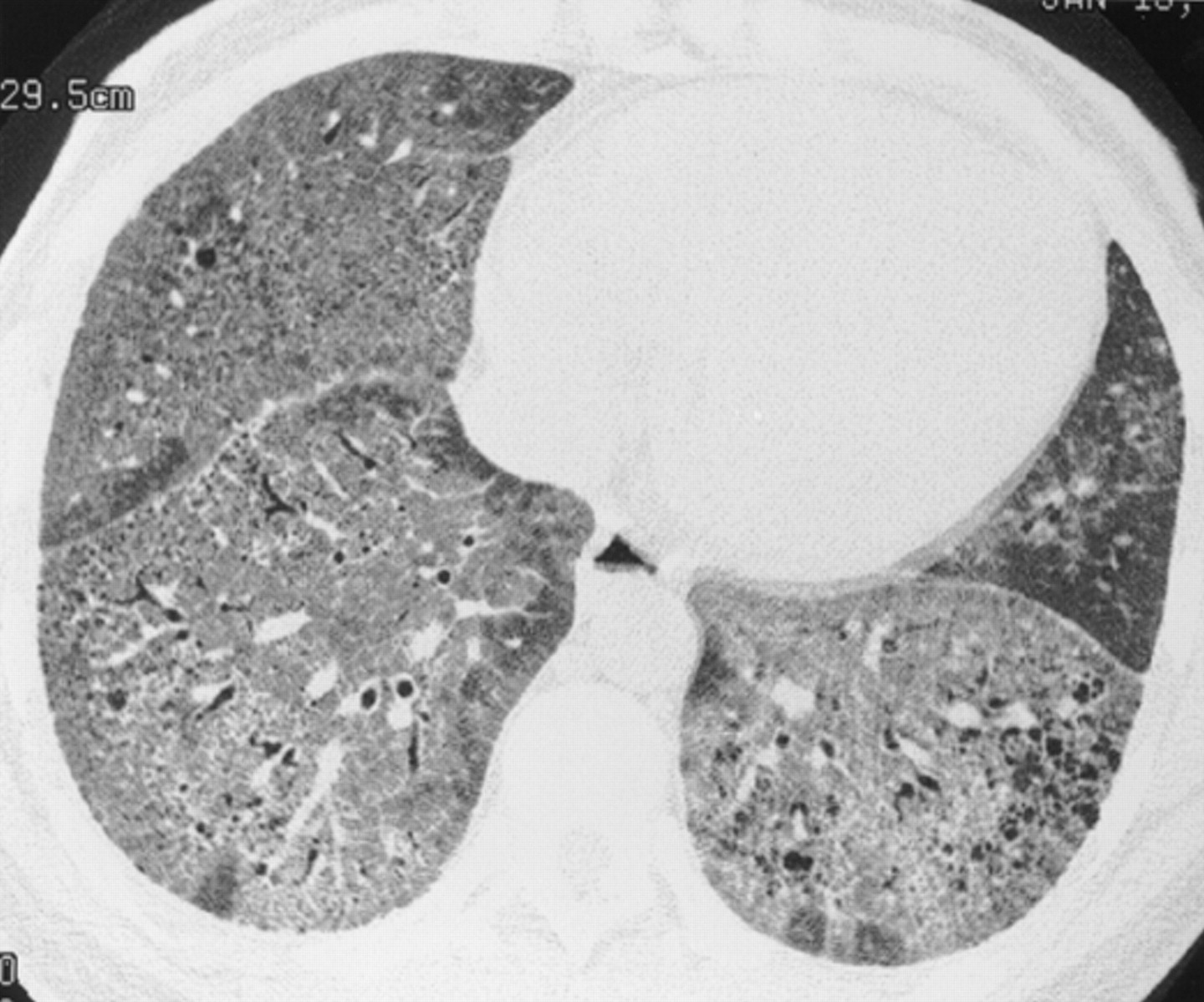
Chronic interstitial pneumonia in a 35 year old woman with SLE. HRCT scan shows extensive ground glass opacities admixed with coarse linear bands and honeycomb cysts. Open lung biopsy confirmed the diagnosis. Ground glass opacities represented a mixture of fibrosis and interstitial mononuclear cell infiltrates. Treatment with corticosteroids, azathioprine, and later cyclophosphamide failed and she subsequently underwent a single lung transplantation.
Data evaluating treatment are sparse. Corticosteroids, immunosuppressive, or cytotoxic agents may be efficacious, but therapeutic trials are lacking.
Acute lupus pneumonitis
Acute lupus pneumonitis presenting as cough, dyspnoea, pleuritic pain, hypoxaemia, and fever occurs in 1–4% of patients with SLE.3 However, there has been some argument about the existence of acute lupus pneumonitis as a specific entity. In one large necropsy series every case of clinically diagnosed lupus pneumonitis could be explained by other factors such as infection, aspiration, cardiac dysfunction, or uraemia.4 This is not surprising given the non-specific findings of diffuse alveolar damage that are associated with acute lupus pneumonitis.
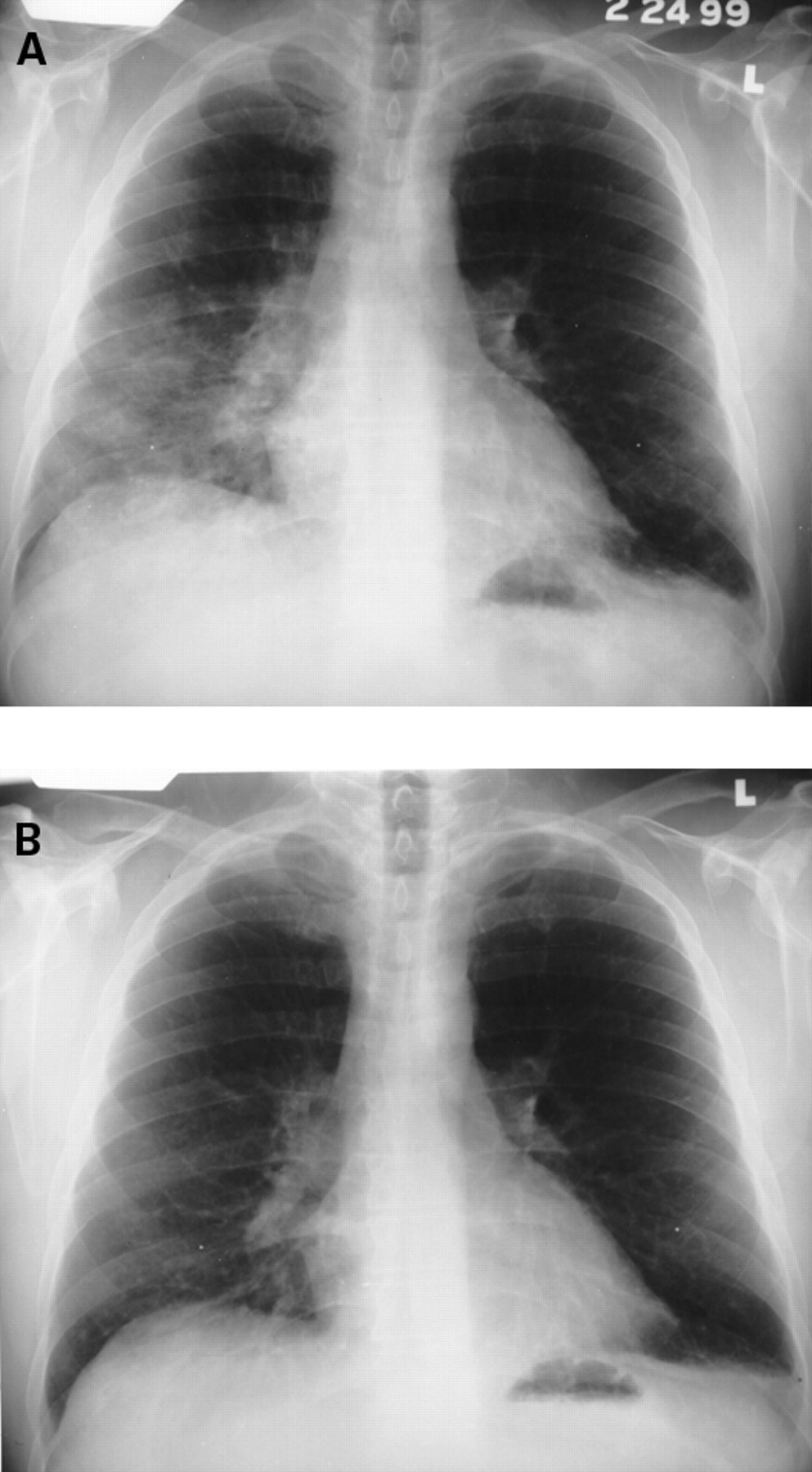
Chest radiographs reveal infiltrates which may be unilateral or bilateral (fig 3). Histological features are non-specific and include alveolar wall damage and necrosis, inflammatory cell infiltration, oedema, haemorrhage, and hyaline membranes.3 A microangiitis involving capillaries, with fibrin thrombi and infiltration with necrotic neutrophils, may be present.24These histological criteria are similar to acute alveolar haemorrhage (see below). Acute lupus pneumonitis is rare and optimal treatment has not been clarified. Favourable responses to corticosteroids are achieved in most patients but the course may be fulminant and occasionally fatal. Immunosuppressive or cytotoxic agents are reserved for corticosteroid-recalcitrant patients.
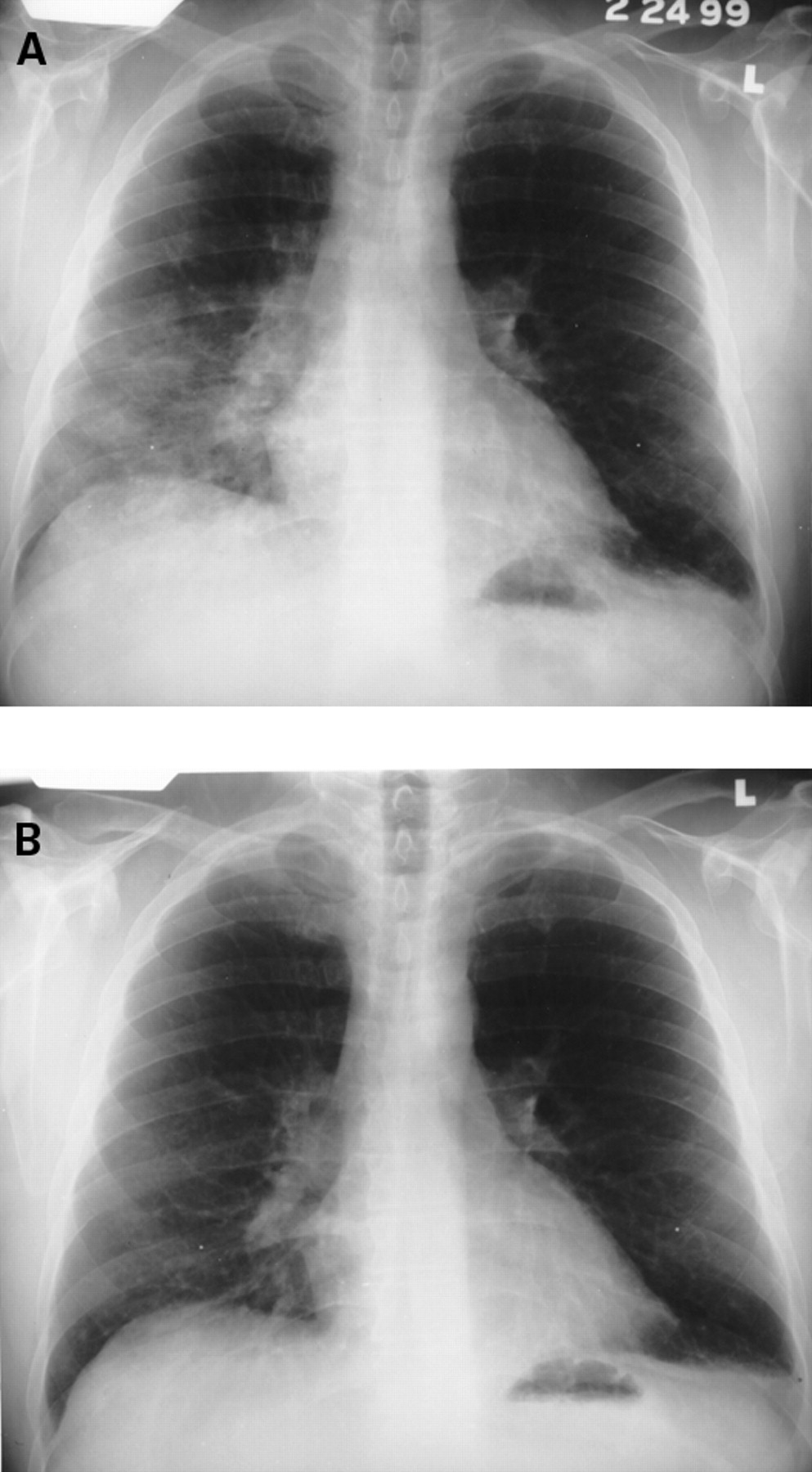
(A) Acute lupus pneumonitis in a 45 year old man. He was treated with corticosteroids and a follow up chest radiograph (B) at four weeks revealed considerable improvement.
Acute alveolar haemorrhage
Pulmonary haemorrhage is a rare but potentially catastrophic complication of SLE.3 Mortality has ranged from 50% to 90%.25 ,26 Clinical features are non-specific but diffuse alveolar infiltrates, hypoxaemia, dyspnoea, and anaemia are characteristic (fig 4).

Acute alveolar haemorrhage. Chest radiograph showing extensive bilateral alveolar infiltrates in a 22 year old woman with SLE, haemoptysis, and anaemia. Reproduced with permission from Keane et al.30
Some of these features may be lacking with minor episodes. Alveolar haemorrhage usually occurs in patients with a known history of SLE, higher titres of circulating anti-DNA antibody, and active extrapulmonary disease.3 However, in up to 20% of cases alveolar haemorrhage may be the initial clinical manifestation of SLE.27 Similar to other pulmonary haemorrhage syndromes, glomerulonephritis is often present in SLE associated alveolar haemorrhage.28 A recent report describes 13 cases of SLE with severe pulmonary haemorrhage and active nephritis; in each case pulmonary haemorrhage occurred after treatment of the nephritis with high dose corticosteroids.29 Lung biopsy specimens demonstrate extensive haemorrhage within alveolar spaces and “capillaritis”; these features are non-specific.24 ,27Deposits of IgG, C3, or immune complexes have been found in up to 50% of patients with alveolar haemorrhage complicating SLE.3 ,24 Because of the potential morbidity we rarely advise surgical (thoracoscopic or open) lung biopsies to diagnosis alveolar haemorrhage. Fibreoptic bronchoscopy with bronchoalveolar lavage (BAL) and transbronchial lung biopsies can be performed with minimal morbidity and is usually adequate to substantiate the diagnosis in patients with suspected alveolar haemorrhage. The presence of gross blood in the airways, serosanguinous BAL fluid, haemosiderin laden macrophages, absence of purulent sputum, and lack of infectious organisms by appropriate stains strongly support the diagnosis of alveolar haemorrhage. Transbronchial lung biopsies may be deferred in critically ill patients with severe alveolar haemorrhage and acute respiratory failure.
Due to the rarity of alveolar haemorrhage complicating SLE, prospective controlled trials to evaluate treatment have not been performed. The treatment of choice is high dose steroids with or without cyclophosphamide; plasmapheresis has been used with anecdotal reports of success.25 In randomised controlled trials plasmapheresis plus prednisone and cyclophosphamide were no more effective than prednisone and cyclophosphamide alone for severe lupus nephritis.3 Plasmapheresis should be reserved for patients with severe alveolar haemorrhage refractory to corticosteroids and cytotoxic agents.30
Pulmonary vascular disease
ACUTE REVERSIBLE HYPOXAEMIA SYNDROME
This syndrome has recently been described in acutely ill patients.31 ,32 The clinical picture consists of hypoxaemia with associated diffusion abnormalities due to an occlusive vasculopathy. There was no association with diffuse parenchymal disease. The combination of activated endothelium and complement activation leads to neutrophil sludging.33 Most cases respond to high dose corticosteroids. Lower dose of corticosteroids in combination with high dose aspirin may improve the pulmonary manifestations but may not be sufficient to control the systemic activity of the disease.2
VASCULITIS
Descriptions of primary pulmonary vascular disease in the absence of parenchymal disease are uncommon. Forty nine cases were reported between 1952 and 1986.34 Demographic features of pulmonary hypertension complicating SLE are similar to those observed in primary pulmonary hypertension (fig 5).3 Raynaud's phenomenon is present in 75% of cases. The pathogenesis of pulmonary hypertension remains obscure, but multiple factors may be operative including pulmonary vasculitis, thrombosis, and pulmonary artery vasoconstriction.3 ,35 The overall prognosis for patients with SLE and pulmonary hypertension is poor with two year mortality exceeding 50%. Treatment is similar to that used in primary pulmonary hypertension with vasodilators being the mainstay of treatment. Some authors have advocated the use of corticosteroids with anecdotal reports of success.36 A recent report advocated the use of combination treatment with immunosuppression, anticoagulation, and vasodilator therapy.37

Chest radiograph from a woman with severe pulmonary hypertension (pulmonary artery pressure 120/60). Note the prominent pulmonary arteries bilaterally with straightening of the left heart border and attenuation of the peripheral vessels. Reproduced with permission from Orens et al.3
THROMBOEMBOLISM
The so-called lupus anticoagulant has been demonstrated in patients with SLE and is associated with an increased risk of intravascular thrombosis.38 Acute and chronic pulmonary emboli are well recognised complications of the anticardiolipin antibody.38 In a review of 842 patients with SLE anticardiolipin antibodies of the IgG or IgM isotype were detected in 204 (24%) and 108 (13%) patients, respectively, and were associated with an increased prevalence of thrombosis compared with patients with negative titres (30% with IgG vs 9% without; 31% with IgM vs 11% without).39 Another review of 390 patients with SLE found that 47% had increased levels of anticardiolipin antibodies but this did not correlate with arterial or venous thrombosis.40The reason for this is unclear but it is possible that only a subset of the anticardiolipin antibodies are pathogenic.40 There was a strong association between the presence of a prolonged partial thromboplastin time and thromboses which may indicate that only those anticardiolipin antibodies with lupus anticoagulant activity are pathogenic.40
Life long anticoagulation may be warranted in patients with recurrent thromboembolic disease.41-44 In addition, intensive treatment with corticosteroids or immunosuppressive agents may be required when anticoagulation alone fails to control thrombosis.3
Airway disease
Bronchiolar disorders rarely complicate SLE. Abnormalities in pulmonary function tests have been detected in up to two thirds of patients with SLE, even in the presence of normal chest radiographs,22 ,23 but severe airways obstruction is rare. In one study pulmonary function tests were performed in 70 non-smoking patients with SLE and a normal chest radiograph and no prior history of asthma or bronchitis.22 Reductions in the FEV1/FVC ratio and maximal mid expiratory flow rate were detected in 5.7% and 24% of patients, respectively, consistent with small airways dysfunction.22 In contrast, FEV1/FVC ratios were normal in all 70 age matched controls without SLE. Another study of 57 consecutive outpatients with SLE documented abnormal pulmonary function tests in 65% with airways obstruction in nine (16%).23 Thus, physiological aberrations are common in SLE but are often asymptomatic. Severe airways obstruction (in the absence of other causes) has rarely been reported. A few cases of cryptogenic organising pneumonia (COP) complicating SLE have been described.45-50 The rarity of COP may reflect, in part, the dearth of surgical lung biopsy specimens in SLE patients with pulmonary processes.51 Corticosteroid treatment is often initiated for presumed “acute lupus pneumonitis”52 but it is possible that some of these cases are COP. In a classical review COP was found on open lung biopsy specimens in one of 12 patients with acute lupus pneumonitis52 and several further reports have confirmed the association of COP and SLE.45 ,46 ,50 Corticosteroid responsiveness has been reported by several authors although progression in spite of immunosuppression with steroids and cyclophosphamide has been described.47-49 ,53 ,54 We believe corticosteroids are preferable as initial treatment, with immunosuppressive or cytotoxic agents being reserved for patients who fail to respond or are intolerant of corticosteroids.54 We are not aware of any published cases of obliterative bronchiolitis complicating SLE. HRCT scans can be useful to aid diagnosis (fig6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Inspiratory HRCT scan of a 43 year old woman with SLE and obliterative bronchiolitis. Faint areas of ground glass opacity are apparent. (B) Expiratory HRCT scan in the same subject with accentuation of the mosaic pattern of ground glass opacity. Reproduced with permission from Lynch et al.99
Infectious complications
Many defects in host defence have been described in SLE, including defects in alveolar macrophage function, chemotactic and phagocytic activity of neutrophils, T cell number and function, delayed hypersensitivity reactions, dendritic cell, B cell, and natural killer (NK) cell function.3 ,55 However, the clinical significance of such defects is unclear. It is more likely that the high rates of infection in SLE are a reflection of the use of immunosuppressive agents than that they are caused by intrinsic immune defects.9 ,56 The risk of infection in the absence of immunosuppression is small. An aggressive diagnostic approach to exclude infection in any patient with SLE presenting with new pulmonary infiltrates is warranted, particularly if they are receiving immunosuppressive therapy.
Diaphragmatic dysfunction
Diaphragmatic dysfunction or “the shrinking lung syndrome” is characterised by dyspnoea, which may be progressive in nature, and the radiological findings of small lung volumes and basilar atelectasis.57-59 It can be difficult to differentiate respiratory muscle weakness from primary parenchymal disease or pleural causes of low lung volumes without the use of invasive studies. The best means of determining the cause of low lung volumes is with the use of oesophageal and gastric pressure measurements, phrenic nerve stimulation, or diaphragmatic electromyography. Few studies have used invasive studies to assess respiratory muscle status in SLE and consequently the true incidence of diaphragmatic dysfunction is not known. Furthermore, the picture can be clouded by the use of systemic corticosteroids which can lead to muscle weakness. Improvement has been reported in individual cases with the use of corticosteroids, inhaled β agonists, and theophylline.58-60
Drug induced SLE
Pleuropulmonary manifestations of drug induced SLE are similar to idiopathic SLE with a similar spectrum of diseases reported.61 ,62 More patients will develop serological evidence of drug induced SLE than will develop clinical evidence of disease.62 They appear to be significantly more common in procainamide induced SLE than in either hydralazine induced SLE or the idiopathic form.62 Most symptoms of drug induced SLE are self-limiting once the drug is discontinued, but occasionally treatment with corticosteroids is required.62 Rechallenge with the offending drug usually results in a recurrence of the disease.62
Pathogenesis
The mechanisms responsible for the development of autoimmune diseases such as SLE are poorly understood. Autoantibody production and polyclonal B and T cell activation are seen, but this polyclonality appears to be restricted to certain autoantigens.63 In animal models of SLE polyclonal activation usually precedes autoantibody production.64-68 Furthermore, there is evidence in both human and murine SLE that autoantibody production results from antigen driven responses.65 ,69 In an animal model of SLE there is evidence of multiple mechanisms of polyclonal T cell activation. Singh and associates showed that SLE prone mice, (NZB × NZW)F1, develop spontaneous T cell activation to several peptides derived from self-IgG and the number of autoantibody augmenting peptides increased as the mice aged, a phenomenon termed “intramolecular determinant spreading”.63 They also showed that a single T cell hybridoma established from a single (NZB × NZW)F1 mouse that had been immunised with one self-IgG peptide was able to recognise several other determinants that had little homology with the immunising peptide, a process termed “determinant degeneracy”.63 This, coupled with recurrent use of T cell determinant sequences among autoantibodies, results in polyclonality that is restricted to certain autoantigens.63
The concept of molecular mimicry has recently received attention as a proposed pathogenic mechanism for autoimmune diseases including SLE.70 Experimental models using the New Zealand mouse have shown that animals with persistent viral infection (RNA or DNA) have accelerated autoimmune responses and develop anti-DNA antibodies.70 Furthermore, the NZW mouse which does not develop autoimmune disease spontaneously, but has the necessary gene(s) to develop anti-DNA antibodies, does so after viral infection.70 Moreover, epidemiological and serological correlations between viral infections and the development of multiple sclerosis, insulin dependent diabetes, and ankylosing spondylitis have been described.71-73 Viruses can induce autoimmunity in several ways such as polyclonal activation, stimulation of cytokine release leading to lymphocyte activation, and by means of a process known as epitope spreading.74-76 Epitope spreading refers to the process by which priming of self-reactive T cells occurs in response to sequestered autoantigens released by virus specific T cell mediated demyelination. Viruses can also contain peptide sequences that mimic “self proteins” and it is this process which is known as molecular mimicry.
It has also been suggested that autoimmune disorders are a result of mutations in the receptors for tumour necrosis factor (TNF) and its related ligands which are important in mediating apoptosis.77 Mutations of the TNF receptor prevent apoptosis leading to a compensatory increase in TNF levels thus exacerbating the inflammatory damage that is typical of complex autoimmune disorders.77 Specifically, mutation in the Fas receptor gene leads to a lymphoproliferative disorder with splenomegaly and signs of autoimmunity at an early age.78 ,79 Fas is a member of the TNF ligand family that plays an important role in lymphocyte apoptosis.77
Role of inflammatory mediators
While autoimmunity is the hallmark of SLE, the development of tissue injury requires the complex interaction of inflammatory cells and mediators and the vascular endothelium. In most instances the tissue inflammation and associated vasculitis involves complement activation, with or without immune complex deposition.80
The complement system is organised into two pathways with C3 occupying a central role. Antibody-antigen interactions result in activation of the classical pathway whereas the alternative pathway is persistently activated at low levels by the interaction of C3 with water.80 C3 is biologically inactive but is cleaved by C3 convertase to yield C3a and C3b. C3b is a potent activator of mast cells and basophils whereas C3a acts as an opsonin on the surface of foreign particles or cells. Both pathways lead to cleavage of C5, thus generating C5a which is a potent chemotactic agent for neutrophils, mononuclear phagocytes, and eosinophils.80 Both the classic and alternative pathways also generate c5b-9 (also known as membrane attack complex (MAC)). MAC is inserted into the membrane of cells ultimately leading to pore formation. Intravascular activation of complement is mediated in a variety of ways, predominantly via antigen-antibody complexes and bacterial products and toxins. Injury to endothelial cells can also lead to activation of complement.
During exacerbations of SLE there is a decrease in serum levels of C3 and C4 in most patients which reflects consumption of complement components and deposition in tissue.81 Chronically depressed levels of complement have also been reported due to factors other than consumption, such as hereditary deficiencies.82It has therefore been proposed that circulating levels of complement degradation products are a more accurate reflection of disease activity.83-87
The vasculitis associated with SLE is most often due to immune complex deposition in the wall of blood vessels.88 This leads to local activation of complement and infiltration of neutrophils with an associated leucocytoclastic vasculitis.81 This is typical of the classic Arthus reaction or experimental serum sickness. An alternative inflammatory vascular injury which does not involve immune complex deposition has been implicated in patients with CNS involvement.33 ,85 ,89 This involves intravascular activation of complement causing neutrophil and platelet sludging with adherence to the endothelium leading to an occlusive vasculopathy. This is similar to the experimental Shwartzman reaction which involves the intradermal injection of endotoxin followed by the intravenous injection of endotoxin.90 Furthermore, the intravascular activation of complement in association with widespread endothelial cell activation can result in diffuse microvascular injury with resultant microinfarction which is the classic pathological finding in patients with SLE with CNS involvement.91-95 The importance of endothelial cell activation and adhesion molecules was seen in a study which showed that ICAM-1 deficiency resulted in a striking improvement in the survival of Faslprmice.96 Furthermore, there is evidence of upregulation of ICAM-1 in skin lesions associated with SLE97 and increased circulating levels of soluble adhesion molecules (E-selectin, sICAM-1, sVCAM-1) have been reported in SLE.98
Conclusions
The pleuropulmonary manifestations of SLE range in severity from the minor pleuritic pain of serositis to the life threatening consequences of pulmonary haemorrhage. Many of the abnormalities have non-specific presentations and require extensive work up to determine the aetiologies. There are few data regarding treatment for most of the immune mediated pulmonary manifestations of SLE. Further controlled trails are warranted to assess the various treatments, although the rarity of some of the manifestations preclude this. Moreover, further investigation is required to understand the pathogenesis of SLE.
Acknowledgments
This work was supported in part by National Institutes of Health grants HL03906 (MPK) and P50HL56402 (JPL).