Article Text

Abstract
BACKGROUND We have consistently argued that mild asthma is an important underlying aetiological factor in patients with severe symptomatic hyperventilation. While hyperventilation has been demonstrated in acute asthma, there have been few studies in mild chronic asthma, and mechanisms are uncertain.
METHODS Twenty three currently asymptomatic chronically asthmatic patients (occasional use of bronchodilators, normal lung function, hyperresponsive to methacholine) were studied and 17 matched normal subjects acted as controls. Ventilation, pattern of breathing, arterial carbon dioxide and oxygen tensions (Paco 2, Pao 2), end tidal Pco 2(Petco 2), standard lung function, airway responsiveness to methacholine, airway inflammation assessed by eosinophils in induced sputum, and psychiatric morbidity (Spielberger STAI-Y and Beck Depression Inventory) were measured.
RESULTS Despite the absence of current asthmatic symptoms, no clinical evidence of hyperventilation, and normal lung function in the patients with asthma, Paco 2 and Petco 2 were significantly (p<0.01) lower in the patients than in the control group (mean (SD) Paco 2 4.96 (0.43) kPa for patients versus 5.27 (0.38) kPa for controls (mean difference 0.31 kPa, 95% confidence interval (CI) 0.06 to 0.56, p<0.02)). Petco 2 was very similar to Paco 2 in both groups (mean (SD) Petco 2 4.89 (0.47) kPa for the patients and 5.28 (0.40) for the controls (mean difference 0.39 kPa, 95% CI 0.12 to 0.66, p<0.01)). There was no significant difference in ventilation or respiratory pattern between the two groups. The reduced Paco 2 in the asthmatic patients correlated significantly with the concentration of methacholine provoking a fall in FEV1 of more than 20% (PC20) (r = 0.56, p<0.01) but not with any aspect of lung function, eosinophil count, or anxiety/depression.
CONCLUSION Mild asymptomatic asthma is not associated with clinically significant hyperventilation but is associated with a significant reduction in both arterial and end tidal Pco 2 which relates to airway hyperresponsiveness rather than to the degree of airway obstruction or mucosal inflammation. Anxiety and depression appear not to be implicated.
- breathing pattern
- hypocapnia
- carbon dioxide
- hyperventilation syndrome
- asthma
Statistics from Altmetric.com
Hyperventilation is well recognised as a concomitant of acute asthma, and studies in which arterial carbon dioxide tension (Paco 2) was related to the severity of acute asthma show clear evidence of significant hyperventilation with only modest reductions in forced expiratory volume in one second (FEV1).1
The mechanisms of hyperventilation in asthma remain uncertain.2 Increased respiratory drive may be caused by stimulation of irritant receptors or C fibres by mucosal inflammation and oedema,3 ,4 or stimulation of stretch receptors related to the increase in end expiratory lung volume.5Hypoxia may stimulate respiration in more severe exacerbations of asthma6 but hyperventilation is not dependent on hypoxia.5 Perception of changing airway resistance may contribute to the increased respiratory drive in asthma and is related to disease severity and frequency of exacerbations, mild stable asthmatics being more sensitive to change than those with severe brittle asthma.7 Psychological factors such as panic, anxiety, and depression8 may exacerbate bronchoconstriction by causing hyperventilation. There is conflicting information about the effect of airway hyperresponsiveness on respiratory control. However, acute asthma is often associated with anxiety, dehydration, infection, and other disturbances which may contribute to lowering of the Pco 2. There is little information about the presence of hyperventilation in patients with chronic mild or moderate asthma in the absence of these possible acute confounding factors.
The aetiological factors which contribute to symptomatic hyperventilation or hyperventilation syndrome remain uncertain.9 ,10 We have consistently argued that a major aetiological factor is very mild and often previously undiagnosed asthma which can lead to a vicious cycle of panic and disability.9 ,11 ,12 For example, we have described a patient who presented with severe hyperventilation and tetany who was subsequently found to have asthma as the sole underlying aetiological factor for hyperventilation.11 In another study12 we investigated a group of patients presenting to our accident and emergency department with a primary diagnosis of hyperventilation. Approximately 80% had good evidence of underlying asthma which had been previously unrecognised in half of them. The association between symptomatic hyperventilation and asthma has been emphasised by others. For example, Demeter et al 13 found an association between asthma and hyperventilation syndrome in 36 out of 38 patients studied. However, the relationship between asthma and hyperventilation is complex and it can be difficult to distinguish true asthma from asthmatic symptoms induced by overbreathing.14 Nevertheless, in the study of hyperventilation in acute asthmatics by McFadden et al, a reduction in FEV1 of approximately 30% was associated with a reduction in Paco 2 to nearly half the normal level,1 well within the range of 1.86–3.86 kPa (14–29 mm Hg) within which hypocapnia can cause symptoms in normal subjects.15 It would therefore be reasonable to propose that mild chronic asthma may be associated with even more marked hyperventilation and hypocapnia than has been described in acute asthma.
We therefore studied a group of patients with mild stable chronic asthma in comparison with a matched control group to determine (1) whether and to what extent hyperventilation is present in this group and (2) if present, whether arterial and/or its equivalent end tidal Pco 2 (Petco2 ) can be found to correlate with any aspect of airway function, airway hyperresponsiveness, airway mucosal inflammation, pattern of breathing, or psychiatric morbidity.
Methods
SUBJECTS
We recruited 23 subjects from our clinical trials unit with a clear history of asthma who were asymptomatic at the time of the study. They all had a history of a positive methacholine challenge test, normal lung function, and were atopic as confirmed by skin prick testing to common allergens. The subject details are shown in table1.
Asthma history and medication of patients with asthma
We also studied 17 control subjects with no history of respiratory or cardiovascular disorders including asthma, allergic rhinitis, exercise wheeze or limitation, or history of childhood chestiness or bronchitis as elicited by a detailed questionnaire. These subjects were matched for age, sex, and height with the asthmatic subjects. Lung function was normal and methacholine challenge was in the normal range (PC20 >8 mg/ml).
All the patients with asthma were young (age range 18–37 years) to reduce the possibility of unrelated disorders. A detailed clinical history was initially obtained. All had a long history of asthma with a time course of 20 years or more in 13 subjects and 10 years or more in a further seven. All subjects had a long history of intermittent bronchodilator use and could be divided into “very mild” with usage of bronchodilators less than once a month (n=7), “mild” with usage once or twice a week (n=10), and “moderate” with usage once or twice a day (n=6). These frequencies represent the average over the previous five years and do not necessarily reflect bronchodilator intake at the time of the study. Many of the subjects used their bronchodilators prophylactically before exercise rather than in response to actual symptoms. Two subjects in the “moderate” group reported occasional nocturnal symptoms but not within seven days of the study. No subject had taken oral or inhaled steroids within six months of the study and seven had never needed steroids of any form. None were taking any form of theophylline or any other medication. Thirteen percent of the patients and 12% of the control subjects were current smokers. A further 26% of patients and 18% of controls were ex-smokers. No subject had a history of any other disorders and no subject gave any history of paraesthesiae, cramps, or cerebral symptoms suggestive of hyperventilation.
The asthmatic patients were asymptomatic at the time of the study and for at least eight hours beforehand, and were not studied if they took, or felt the need to take, bronchodilators in the previous eight hours either at rest or during exercise, or if they had experienced any wheeze, shortness of breath, or chest discomfort over the same time period. These questions were asked on a number of occasions and in a number of different ways during the course of the study to ensure truthful replies. Subjects were rejected if they reported any acute allergic or infective exacerbations of their asthma during the previous eight weeks. All subjects avoided caffeine for at least eight hours before the study, which was completed in one day for each subject.
Ethical permission was obtained from King's Healthcare Research ethics committee and subjects gave informed signed consent.
PROTOCOL
We performed the measurements described below in the following order: psychiatric questionnaires; ventilation and breathing pattern; arterial blood gas tensions; lung function; methacholine challenge; eosinophil count from induced sputum. All measurements were performed within a four hour period on one day. Care was taken to ensure that both groups were studied with an identical protocol in the order described. Skin prick testing for common allergens was performed on arrival.
Psychiatric assessment
Subjects completed a range of medical and psychological questionnaires. Psychiatric rating scales are established methods of screening for psychiatric disorder and a number of self-rating scales have been validated for depression and anxiety. They can be useful in differential diagnosis and have been used to screen for such disorders in chronic disease. The self-rating scales used in the present study were the Spielberger State Trait Anxienty Inventory (STAI)16 and the Beck Depression Inventory (BDI).17
Ventilation and pattern of breathing
Ventilation was measured via a mouthpiece with a nose clip using a heated pneumotachograph (PK Morgan, Chatham, Kent, UK) with the subject in the sitting position. Subjects breathed quietly for 15 minutes and then five minutes of ventilation was recorded and averaged by a computer system (Systematica Ltd, UK) in which the pattern of breathing and Petco 2 were measured in real time. Minute ventilation (V˙), tidal volume (Vt), inspiratory time (Ti), expiratory time (Te), cycle duration (Tt), mean inspiratory flow (Vt/Ti), and respiratory frequency (f) were recorded for each breath. Respired air was sampled continuously at the mouth and analysed for Pco 2 by a P K Morgan capnograph 901-MK2 (Chatham, Kent, UK). Petco 2 was measured by the computer system, which also measured the slope of the end tidal plateau and the timing of the inspiratory and expiratory phases of each breath from the Pco 2 profile to allow removal of invalid breaths. A maximum of 10% of breaths were removed by such editing procedures. The mean of the remaining values provided the measures of Petco 2 in the subsequent analysis.
Arterial blood gas tensions
Arterial blood gas tensions were measured during ongoing ventilatory recording about two minutes after measurement of the respiratory pattern. Arterial blood was sampled from the non-dominant radial artery under local anaesthetic over at least 30 seconds. The sample was analysed immediately (AVL 995, AVL Medical Instruments AG, Schaffhausen, Switzerland) to give Paco 2, Pao 2, pH, HCO3 –, and base excess. Petco 2 was also measured during the arterial sampling (Petco2 (s)) and provided a means of validating the end tidal measurements.
LUNG FUNCTION
Lung function variables measured included FEV1 and forced vital capacity (FVC) (Vitalograph, Buckingham, UK), peak expiratory flow rate (PEF) (Wright peak flow meter), carbon monoxide transfer factor (Tlco) and carbon monoxide transfer coefficient (Kco) (P K Morgan Transfertest Model C), residual volume (RV) and total lung capacity (TLC) (P K Morgan body plethysmograph). All were expressed as percentage predicted for age and height.
BRONCHIAL REACTIVITY
Bronchial reactivity to methacholine was performed after lung function measurements (breath activated dosimeter, Mefar, Italy) and the concentration (mg/ml) that induced a fall in FEV1 of 20% from post saline inhalation (PC20 FEV1) was calculated according to a standard protocol.18 The maximum concentration of methacholine used was 32 mg/ml. Criteria for entry into the asthmatic group were a PC20 of <8 mg/ml and for the control group >8 mg/ml.
AIRWAY INFLAMMATION
Airway inflammation was assessed by measuring the percentage of eosinophils in induced sputum. Fifteen minutes after inhalation of salbutamol 200 μg via metered dose inhaler, hypertonic saline at concentrations of 3%, 4%, 5% were inhaled for seven minutes, each followed by spirometric tests for safety.19 The subjects were asked to rinse their mouth with water before and after each nebulisation. Sputum was expectorated every seven minutes and collected. Sputum plugs were selected and mixed with a volume of 0.1% dithioreitol (Sputolysin Reagent, Calbiochem, San Diego, USA) four times the sputum weight, vortexed, and left to homogenise on a bench rocker for 15 minutes at room temperature.20 Further dilution with the same volume of phosphate buffered saline (PBS) preceded sample filtration through a 48 μm nylon gauze. The total cell count was adjusted with PBS to give a concentration of 200 000 cells/ml. Aliquots of 100 μg were applied to cytospin slides and stained using Wright's Giemsa (Sigma, Poole, UK) for differential cell counts which were performed blind on 400 non-squamous cells and eosinophils expressed as a percentage of the total cell count.
ANALYSIS OF DATA
Values for PC20FEV1 were log transformed to allow statistical analysis and presented as a geometric mean. All other data are presented as mean with standard deviation (SD) or standard error (SE). Ventilation, Petco 2, blood gas tensions, and lung function variables were compared between groups using the Student's t test. Mann-Whitney tests were used to compare PC20, eosinophil counts, and psychological scores. Parametric (Pearson) or non-parametric (Spearman rank correlation) regression analysis was used to study the relationship between ventilation or Pco 2 and lung function, airway reactivity, airway inflammation, and psychological scores. A p value of <0.05 was considered to be significant except for the correlation matrix when a value of <0.01 was used.
Results
Twenty eight variables were measured; a comparison of the mean values of selected variables between the two groups is shown in table2. Representative variables were selected from each group to give a broad picture of the similarities and differences between the groups.
Comparison of selected variables between groups.
All lung function was in the normal range for both groups. There was no significant difference between the groups for FEV1, FEV1/VC, or gas transfer. PEF was significantly lower in patients than in controls (97.8 (2.8)% predicted vs 107.1 (3.2)% predicted; p<0.05). RV was significantly higher in patients than in controls (108.3 (6.6)% predicted vs 86.6 (5.5)% predicted; p<0.02). The geometric mean PC20 FEV1 for the patients was 1.1 mg/ml. Eleven control subjects had a PC20FEV1 of more than 32 mg/ml, the highest concentration used; the geometric mean PC20FEV1 of the remaining six was 12.5 mg/ml (range 8.1–20.5 mg/ml).
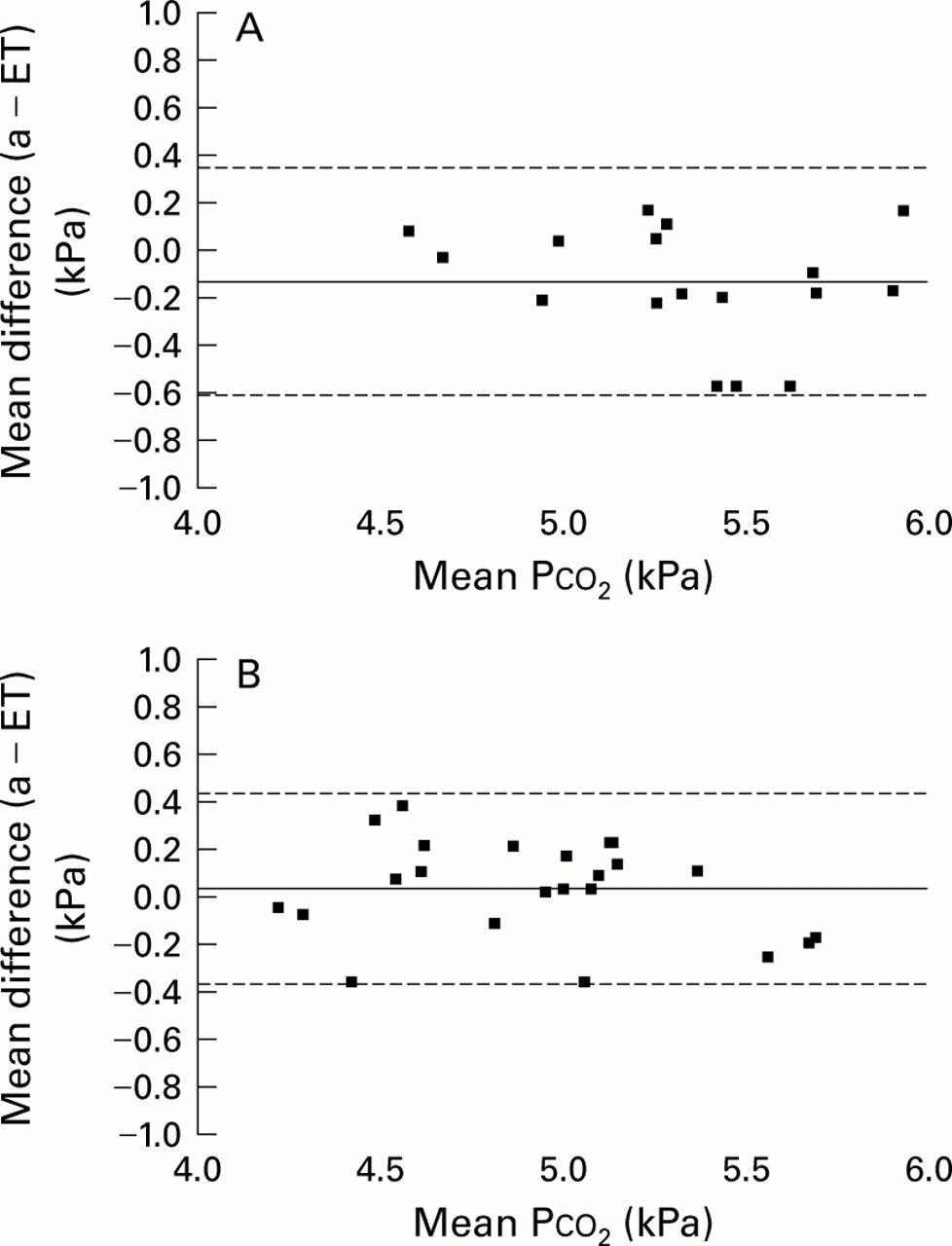
Paco 2 and all measures ofPetco2 were significantly lower in the patients than in the control group with mean (SE) Paco 24.96 (0.09) kPa and 5.27 (0.09) kPa, respectively (mean difference 0.31 kPa (95% CI 0.06 to 0.56), p<0.02). Equivalent values for Petco 2 were 4.89 (0.09) kPa and 5.28 (0.09) kPa, respectively (mean difference 0.39 kPa (95% CI 0.12 to 0.66), p<0.01). Values of Petco2 (s) were very close to the arterial values with a correlation coefficient of 0.91 (p<0.001) and a mean difference of 0.04 (0.04) kPa in the patients, supporting the validity of end tidal measurements in these patients. This compares with a difference of –0.13 (0.05) kPa in the controls. Bland and Altman plots with limits of agreement for the two groups are shown in fig 1.21 The slope of the alveolar plateau was non-significantly greater in the patients with asthma than in the controls (0.47 (0.09) kPa/s vs 0.37 (0.04) kPa/s). There was a significant difference (p<0.01) between male and female subjects for mean Paco 2 in the control group (5.48 (0.29) kPa vs 4.95 (0.28) kPa) but not in the patient group (5.08 (0.45) kPa vs 4.83 (0.33) kPa). The male/female ratio was similar in the two groups.
{kind=link}
Bland and Altman plots for (A) controls and (B) patients with asthma showing mean Paco 2 and Petco 2 values on the x axis and the difference between the two on the y axis. The mean values for individual subjects are shown; the solid horizontal lines represent the mean differences and the broken lines represent the limits of agreement.
There was no significant difference between the two groups for other blood gas measurements and ventilatory variables. The reducedPco2 in the patient group was associated with a slightly, but not significantly, greater ventilation and respiratory frequency with equivalent shortening of Ti, Te, and Tt (table 2). Pao 2 was higher in the patients than in the control subjects, which was consistent with the reduced Paco 2. Base excess was negative in both groups.
Four control subjects produced poor sputum samples that could not be analysed. The sputum eosinophil count in the patients was significantly higher (p<0.01) than in the controls (median 3.0 (range 0–27) vs 0 (range 0–3.2), n=13).
The patients had a significantly greater anxiety state score than the controls (p<0.02) despite the absence of clinical evidence of anxiety, but both scores were still within the normal range (25–35 for both state and trait). The BDI score for individuals was also within the normal range (<10) and there was no significant difference between the two groups.
The patient group was divided into those with bronchodilator use of less than once a month, those with bronchodilator use of less than once a day, and those using bronchodilators more often than once a day. These groups were separately analysed for a limited but representative selection of variables (table 3). No significant difference was found for any variable between any of the groups, and there were no obvious trends as asthma worsened apart from a non-significantly higher anxiety score for the more severe subgroups.
Mean (SD) characteristics of patients according to bronchodilator use: very mild (⩽1/month), mild (<1/day) and moderate (⩾1/day)
Correlation coefficients for selected variables in the patients are shown in table 4. There was a significant correlation between PC20 and Paco 2 (p<0.01) and a slightly lower correlation (p<0.02) between PC20 andPetco2 , but there were no equivalent correlations in the controls. Neither Pco 2 nor PC20 correlated with any other measure of baseline airway function or inflammation. There was no relationship between the STAI scores and any measure of baseline lung function, airway hyperresponsiveness, or airway inflammation.
Correlation coefficients between selected variables in patients with asthma
Eight of the 17 control subjects were positive to common allergens on skin prick testing. The atopic and non-atopic controls were analysed as a single group as no differences were found between any variables measured.
Discussion
This study illustrates the value of studying respiratory control and searching for small but significant changes in patients with very mild lung disease, rather than attempting to study more severe disease in which it is often difficult to control for extraneous factors. We have shown that uncomplicated mild and moderate chronic asthma is not associated with clinically significant hyperventilation, but we have demonstrated a small but statistically significant reduction in both Paco 2 and Petco2 .
Although all of our asthmatic subjects were symptomatic at the time of the study and for at least eight hours previously, the reported severity of the asthma in the different subjects when considered over a long period of time varied from bronchodilator usage of less than once a month to usage on a daily basis, so we divided the patients into three subgroups on this basis corresponding roughly to very mild, mild, and moderate asthma. Unfortunately, this grouping was not able to distinguish between subjects who required bronchodilators for an actual attack of asthma and those who took their inhalers prophylactically—for example, before exercise. There was no consistent trend in any variable as severity increased apart from higher anxiety scores in the two more severe groups, and there was no significant difference in any variable between these three subgroups. There was an equivalent reduction in both Paco 2 andPetco2 in each group.
No subject reported any symptoms suggestive of hypocapnia or hyperventilation, and we did not expect such symptoms as we specifically wished to study fit young subjects with only mild asthma uncomplicated by any clinical evidence of psychiatric or other problems. For the same reason, we did not perform any stress tests for hyperventilation9 although the results of these may have been of interest.
These results do not provide a ready explanation for the extreme symptomatic hyperventilation that we have observed in some of our patients,11 ,12 and do not show that patients with mild chronic asthma have marked hypocapnia at rest as we had predicted. Other factors need to be sought to explain such a linkage in some patients. Nevertheless, in this study the range of Paco 2 in the patients with asthma was large, extending from 4.19 to 5.59 kPa (31.4 to 41.9 mm Hg). Three of the 23 asthmatic subjects had a Paco 2 value of less than 4.27 kPa (32 mm Hg), very close to the upper limit for induction of symptoms by voluntary hyperventilation in normal subjects,15 and seven had values below 4.67 kPa (35 mm Hg), the lower limit of the accepted normal range forPetco2 . This contrasts with the control group in which only two subjects had Paco 2 values below 4.67 kPa and these were only 0.05 and 0.01 kPa, respectively, below the lower limit of the normal range. These results are only applicable to subjects breathing on a mouthpiece which can itself influence breathing22 and which may have prevented more impressive hyperventilation when the subjects were uninstrumented outside the laboratory. We are currently restudying this using our new ambulatory capnograph in the same subject group during activities of daily living.23
It is possible that a key factor in determining whether asthma induces hyperventilation sufficient to induce symptoms is prior awareness of the diagnosis of asthma, and such lack of a previous diagnosis was present in the case study described by Gardner et al 11 and in half of our patients presenting to the accident and emergency department with hyperventilation.12About 80% of the patients in the latter study also had evidence of mild anxiety on questionnaires and it is reasonable to propose that dyspnoea and chest tightness which occurs suddenly and without an explanation is a potent source of anxiety and panic leading to hyperventilation, exacerbated by the tendency of patients when faced with unexplained symptoms to take large breaths, although demonstrable psychiatric morbidity was remarkably absent or minimal in many of the patients with symptomatic hyperventilation whom we have previously described.24 ,25 In the present study the anxiety score was significantly higher in the asthma patients than in the controls but was still within the normal range. The relationship between asthma, hyperventilation, and anxiety requires further study.
This study shows a small but significant reduction in both Paco 2 and Petco2 in patients with asthma and suggests that such a reduction is fundamental to the pathophysiology of asthma and is not an artefact or a paraphenomenon. Nevertheless, an explanation is required for our finding of a lower Paco 2 in the asthma patients without evidence of significant hyperventilation. It should first be emphasised that Paco 2 is dependent on alveolar, not inspired or expired, ventilation and we had no measurements of dead space or alveolar ventilation in our study. Although none of the patterning variables was significantly different, minute ventilation and respiratory frequency were slightly greater and both Tiand Te were shorter in the patients than in the controls; these differences may have become significant with a larger number of subjects. We do not believe that our finding of a lower arterial and end tidal Pco 2 in the patients with asthma was related to methodological differences between the way in which the two groups were studied. Extreme care was taken to ensure that both groups were studied with exactly the same protocol and the methacholine challenge and induced sputum measurements were always performed after the measurements of Pco 2 and thus could not have influenced the results.
In searching for other mechanisms which may have led to the increase in alveolar ventilation in the patients, it may be of significance that there was significant correlation of individual values of Petco2 and Paco 2 with corresponding values of PC20 and it is conceivable that the mucosal inflammation and oedema associated with asthma resulted in a reduction in the dead space, contributing to an increase in the alveolar component of ventilation. However, dead space probably does not fall in asthma although it does fail to increase normally with inspiration.26 Neither group were hypoxic and this cannot provide an explanation. RV was significantly increased in the asthmatic patients and increased functional residual capacity (FRC) may be a factor in the hyperventilation associated with acute bronchospasm,5 ,27 probably mediated by stimulation of lung or chest wall mechanoreceptors, but it is difficult to understand how this could affect alveolar and not inspired ventilation. In this study reduction of Pco2 could not be attributed to any aspect of medication as the asthmatic subjects had not taken bronchodilators for at least eight hours before the study and none had taken steroids or other medication for many months.
Anxiety can induce hyperventilation and our asthmatic subjects had a significantly higher anxiety state score than the control group, with non-significantly higher scores in the more severe patients than in those with milder asthma. However, all psychiatric scores were within the normal range, and there was no suggestion of correlation between any of these scores and either Paco 2 orPetco2 . The significant correlation between STAI and BDI scores is of interest but is not surprising.
The highly significant correlation between PC20 and Paco 2 and the less significant (p<0.05) correlation with Petco2 was striking. These are both independent variables, and this result should be contrasted with the complete lack of correlation with the markers of inflammation. While correlation matrices such as this can produce random significant values and the results should thus be interpreted with caution, it is reassuring that the only other significant correlations were those between related variables such as PEF and FEV1, V˙ and mean inspiratory flow, f and Vt, and the psychiatric scores against each other. The ventilatory responses to bronchoconstriction induced by bronchial challenge in the literature are variable3 ,4 ,28 ,29 and occur before the reduction in FEV1 30 and when bronchoconstriction is prevented by terbutaline inhalation, suggesting that increased respiratory drive may be induced by direct stimulation of vagal airway receptors. Inflammatory mediators may stimulate respiration by the same mechanism, but our results suggest that these did not play a part in the reduction of Paco 2 in our patients.
We have shown a relationship between arterial and end tidal Paco 2 and bronchial hyperresponsiveness but not with airway inflammation as measured by induced sputum eosinophil count. Both measures are widely used to assess the potential benefits of new asthma treatments19 ,20 but their relationship remains unclear. Allergen challenge induces an influx of eosinophils into the airways and a corresponding increase in airway hyperresponsiveness31 and may induce hyperventilation by stimulation of vagal afferents.4 Inflammatory mediators may contribute to the lowering of the threshold for stimulation of vagal afferent nerve endings, and a similar decrease in threshold may contribute to airway hyperresponsiveness in asthma. However, airway hyperresponsiveness is present even in the absence of inflammation32 and persists after steroid therapy. In our study, in agreement with others, we found no relationship between airway hyperresponsiveness and airway inflammation in the asthmatic group.
This study confirms that end tidal measurements of Pco 2 are accurate reflections of arterial Pco 2 in mild and moderate asthma, the mean alveolar–arterial difference for the asthma patients being 0.04 kPa compared with 0.13 kPa for the controls, despite a small but expected increase in the slope of the alveolar plateau in the patients. The Bland and Altman plots (fig 1) show no consistent change in this difference across the range of values obtained, and show very similar plots for both controls and asthmatics. These results support another study showing a good association between arterial and end tidal values when FEV1 was over 80% predicted.33 We would argue that end tidal measurements can be used with confidence in mild and moderate asthmatic patients as accurate reflections of arterial measurements.
In summary, we were not able to demonstrate clinically significant hyperventilation in our patients with mild and moderate chronic asthma, but these patients did have a statistically significant reduction in both arterial and end tidal Pco2 unrelated to lung function, airway inflammation, or measures of psychiatric abnormality, but associated with airway hyperresponsiveness. A small degree of hypocapnia may thus be intrinsic to the increase in airway smooth muscles contractility in asthma, but additional factors may also be involved in more severe acute exacerbations of asthma. This study will provide the basis for further studies both in mild asthmatics using non-invasive measurements by ambulatory techniques outside the laboratory, and in patients with severe hyperventilation associated with mild asthma.
Acknowledgments
The authors would like to thank the Wellcome Trust for financial support and the Guy's, King's and St Thomas' School of Medicine for laboratory facilities.