Article Text

Statistics from Altmetric.com
Airway inflammation in chronic obstructive pulmonary disease (COPD) is currently the subject of rapidly increasing research interest, investigating both the nature of the inflammatory cells and the cytokines present. The data are being used to define and assess the severity, cause, prognosis, and response to treatment of this disease. The collection of spontaneous sputum or the induction of sputum has been used to study inflammation in the larger airways. It remains unclear, however, how these data relate to the pathological processes at the site of airflow obstruction (the small airways and alveolar region). Nevertheless, even the interpretation of the results from the large airways depends on the many factors that will influence the inflammation.
The initial process of sputum collection, sample processing, and the performance of assays in the biological fluids being tested may all influence the results. In addition, airways inflammation may be altered by the patient's clinical state, current treatment, and the nature of the disease (asthma, COPD, or bronchiectasis). Other factors including smoking, α1-antitrypsin (AAT) deficiency, and bacterial colonisation may also affect airways inflammation. Despite these many factors that may influence airways inflammation, many papers currently in the literature do not address them. The purpose of this review is to indicate the influence of these factors on airways inflammation to assist interpretation of the data currently in the literature.
Pathophysiology
EOSINOPHIL
Airway inflammation in asthma is classically related to the eosinophil. Eosinophil granules contain a variety of mediators capable of causing inflammatory damage to the airway epithelium including eosinophil peroxidase (EPO), major basic protein (MBP), eosinophil cationic protein (ECP), metalloproteinases, platelet activating factor, and cysteinyl leukotrienes.1 In addition, the immunological basis of asthma is distinct and reflected in the nature of the cytokines released into the airway.
However, there is some overlap with COPD which may reflect the fact that asthma can lead to fixed airflow obstruction or that patients with COPD may have coincidental asthma.2 Indeed, some recent studies have indicated that sputum eosinophilia is a predictor of steroid responsive airflow obstruction3 ,4 and thus may identify the chronic asthma component or a subtype of the COPD syndrome.
NEUTROPHIL
In contrast, however, the neutrophil is thought to be the major cell involved in the airways of patients with COPD and those with bronchiectasis, although it has also been implicated in the later irreversible phase of asthma. In neutrophilic inflammation in the bronchial tree there is a complex interaction of many mediators including chemoattractants and cytokines that regulate gene transcription, adhesion molecule expression, the processes of cell migration, their activation and degranulation.
PROTEASES
The recruited neutrophil releases proteolytic enzymes including neutrophil elastase, cathepsin G, and matrix metalloproteinases. The importance of these enzymes—and, specifically, neutrophil elastase—has been demonstrated by both in vitro and in vivo studies. Neutrophil elastase can produce most of the pathological features of chronic bronchitis and emphysema, and promote bacterial colonisation as seen in patients with bronchiectasis. More specifically, neutrophil elastase can cause epithelial damage,5 reduce ciliary beat frequency,6 produce mucous gland hyperplasia,7 stimulate mucus secretion,8inactivate many critical lung host defences,9 ,10 and damage interstitial tissue11 leading to pathological changes that cause human alveolar destruction.
ANTI-PROTEINASES
In order for neutrophil elastase to have these effects, the elastase released from the activated neutrophil has to overcome the naturally occurring inhibitors such as secretory leukoprotease inhibitor (SLPI) and AAT. SLPI is generally thought to be the main inhibitor in the airways12 and hence protects against bronchial disease, whereas AAT is thought to be of importance predominantly in the lower airways13 and is thus more relevant to alveolar disease.
CHEMOATTRACTANTS
The major neutrophil chemoattractants in patients with COPD and bronchiectasis are believed to be interleukin 8 (IL-8) and leukotriene B4 (LTB4).14 ,15IL-8 is a 16 kDa protein that is a member of the C-X-C family of cytokines. It is made and released by bronchial epithelial cells,16 monocytes/macrophages,17 and even the neutrophils themselves.18 Regulation of IL-8 production in the lung is complex but recent studies have started to determine the processes involved. It is known that tumour necrosis factor-α (TNF-α) can increase the expression of the IL-8 gene,19 and lung secretions from patients with COPD and bronchiectasis contain TNF-α.20 ,21 Furthermore, recent work has indicated that cigarette smoke can stimulate epithelial cells to release IL-822 via the transcription factor NfκB23 ,24 and this may be the mechanism that leads to neutrophil recruitment to the lungs of smokers. In addition, bacteria can also induce IL-8 expression by epithelial cells19 ,25but at present it is uncertain whether the effect of bacterial products on IL-8 expression is direct or the result of initial TNF-α secretion. IL-8 levels are raised in patients with COPD and patients with bronchiectasis26-29 and correlate with myeloperoxidase (MPO) released from activated neutrophils.26 ,27 ,29 This relationship between IL-8 and a neutrophil marker supports the concept that IL-8 is an important lung chemoattractant for neutrophils.
LTB4 is a biologically active fatty acid synthesised in myeloid cells from arachidonic acid. It was isolated and purified from neutrophils in 197830 but was subsequently chemically synthesised and its ability to activate neutrophils was shown in 1980.31 LTB4 is now known to be a major chemotactic product of activated neutrophils31 and macrophages.32 LTB4 induces recruitment and activation of neutrophils as well as monocytes, eosinophils, and lymphocytes. It promotes chemotaxis by increasing neutrophil adherence to venule walls via the integrin Mac-1 and its receptor ICAM-1.33
The cytokine TNF-α has been found to be increased in the airways of patients with COPD20 and bronchiectasis.21 It is produced by activated macrophages, mast cells, and other inflammatory cells34-37 as well as by airway epithelial cells.38 TNF-α induces airway hyperresponsiveness,39 may promote IL-8 production by epithelial cells40 and neutrophils,41 is chemotactic for neutrophils and eosinophils both directly and by activation of adhesion molecules on endothelial cells,42 ,43 and promotes vascular leakage.44However, in vivo it has been shown to inhibit neutrophil chemotaxis45 and therefore its role in lung inflammation is complex and requires further study.
There are a number of other chemoattractants that could play a role in the airways including C5a generated from complement activation46; modified AAT47; leucocyte elastase-inhibitor complexes48; protein and peptide components of damaged extracellular matrix such as collagen,49 elastin50 and laminin51; antigen-antibody complexes52; bacterial factors including the formyl peptides such as fMLP53 and lipopolysaccharide or endotoxin which can prime neutrophil functions including the chemotactic responses.54
A significant proportion, however, of the chemotactic activity of lung secretions in patients with COPD and bronchiectasis can be attributed to IL-8 and LTB4. Studies by Mikami et al 14 have shown that up to 43% of the chemotactic activity was dependent on IL-8 and 27% on LTB4. It was further shown that the chemoattractants worked in an additive way for neutrophil recruitment.14 However, these workers also emphasised that some of the chemotactic activity of the lung secretion was due to a protein or proteins that remain to be identified. This is particularly true in patients with bronchiectasis who usually have marked bronchial inflammation associated with significant neutrophil influx. Other potential chemoattractants listed above may play a role and cells migrating into tissues may thus encounter multiple chemoattractant signals. Foxman et al 55 have shown that neutrophils can even migrate “down” a local chemoattractant gradient in response to a distal gradient of a different chemoattractant. In addition, cells can migrate effectively to a secondary distant agonist after migrating “up” a primary gradient into a saturating non-orienting concentration of the initial attractant. These mechanisms of navigation from one gradient to another in complex chemoattractant fields are likely to be necessary for successful localisation and phagocytosis within sites of inflammation.55
The relationship between chemoattractants and neutrophils may be further complicated since neutrophil activation can lead to the release of both IL-818 and LTB4 31 that may enhance further neutrophil recruitment. Release of elastase from the neutrophil may also stimulate epithelial cells to produce more IL-816 and macrophages to release LTB4,32 while at the same time reducing production of its own natural inhibitor, SLPI,56 thereby perpetuating its function. Furthermore, impairment of host defences by elastase could facilitate bacterial colonisation and endotoxin release from bacteria can also provide a further potential mechanism for neutrophil recruitment by stimulating production of IL-8 by epithelial cells.19 The understanding of these complex interrelationships and the role of each component awaits in vivo studies with specific agonists and antagonists.
RELATIONSHIP BETWEEN ELASTASE ACTIVITY AND OTHER INFLAMMATORY MARKERS
Neutrophil elastase is thought to be of major importance in the pathogenesis of airways disease in COPD and bronchiectasis. Studies from spontaneous sputum have shown relationships between neutrophil elastase activity and several features of airways disease including the following: neutrophil influx; chemoattractants (IL-8 and LTB4); SLPI; and protein leakage (as determined by sputum/serum albumin ratio).57 The close correlation between neutrophil elastase and neutrophil influx would be expected as it is stored within the primary granules of the neutrophil and would be released simultaneously with other granule constituents such as MPO following degranulation. Similarly, sputum elastase activity relates closely to the chemoattractants IL-8 and LTB4, probably because these cytokines are responsible for neutrophil (and hence elastase) recruitment. However, as indicated above, elastase can result in IL-8 production by epithelial cells and LTB4 release by macrophages which may partly explain the relationship. Elastase is related inversely to SLPI concentration, probably by directly suppressing its release,56 although a low SLPI concentration would, of course, facilitate continued activity of the enzyme. Finally, as elastase activity is increased above 50 nM, protein leakage also increases, although the exact mechanism is currently unknown. However, previous studies in patients with bronchiectasis have shown that antibiotic treatment reduces the bacterial load and neutrophil influx leading to a rapid reduction in protein leakage as sputum purulence and elastase activity disappear.58 The rapidity of this response would suggest that airway leakage is not a direct effect of major airways damage or epithelial cell destruction. The leakage may therefore reflect an effect on the tight junction between epithelial cells which can be mediated by elastase59 or the simultaneous release of vasoactive mediators such as NK-160 in the airway during inflammation.
Disease process
Induced sputum samples have confirmed the presence of increased concentrations of products derived from neutrophil granules (MPO, human neutrophil lipocalin (HNL)) and from eosinophils (EPO, ECP) in patients with asthma or COPD compared with normal individuals.61Although MPO and HNL were significantly higher in patients with COPD than in those with asthma, the concentrations of ECP and EPO were similar in the two groups of patients indicating a degree of eosinophil influx in addition to neutrophil influx in COPD. The cytokines TNF-α and IL-8 were also increased in both conditions, although only IL-8 was significantly higher in COPD than in asthma.20 Other inflammatory mediators have also been found to be increased in induced sputum, including the tachykinin substance P in individuals with asthma and chronic bronchitis.62
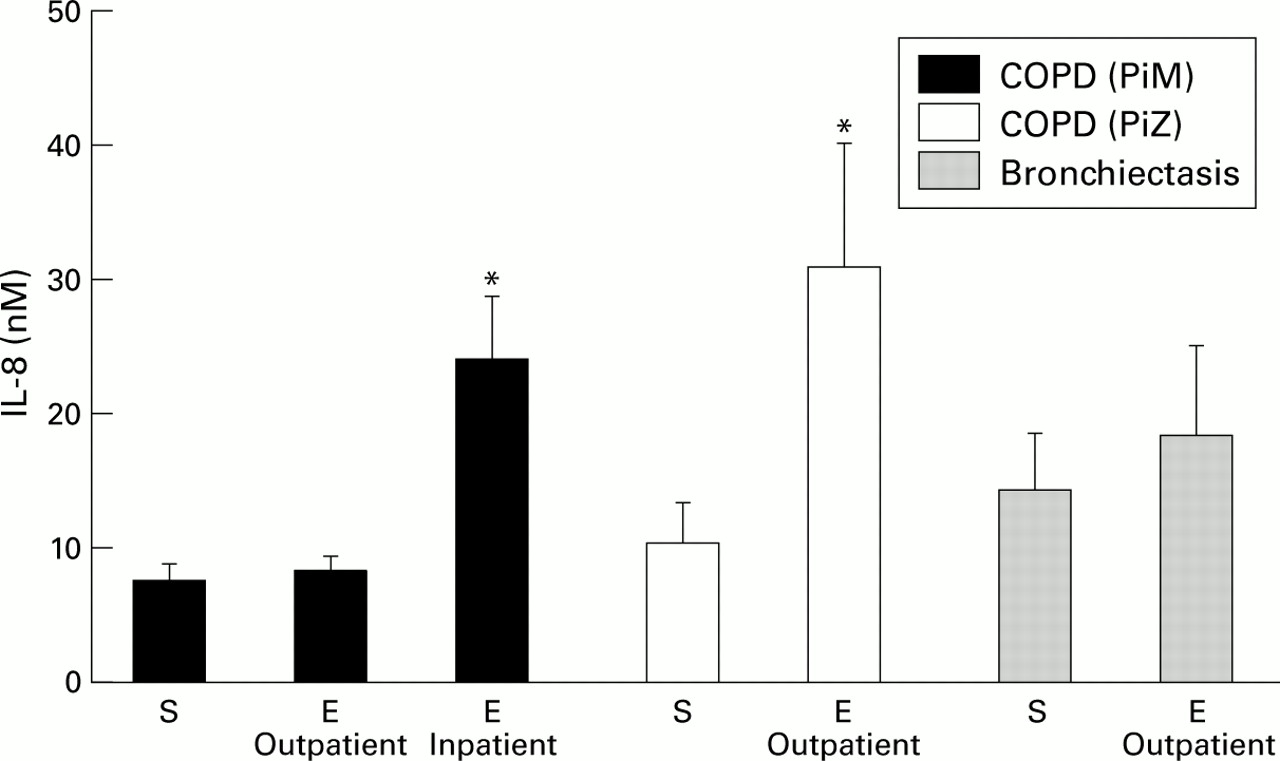
Patients with bronchiectasis are characterised by major airways inflammation thought to be driven by persistent bacterial colonisation.63 Such patients have been shown to have significant neutrophil influx with raised MPO63 and readily detectable elastase activity,64 cathepsin B,65 proteinase 3,66 increased chemoattractants (IL-8, TNF-α, IL-1β, and LTB4),21 increased endothelin-1 (a potent bronchoconstrictor with mitogenic activity for airway smooth muscle),67 and increased albumin protein leakage.64 Patients with COPD also have airways inflammation, but to a lesser degree.68 ,69 This is illustrated in fig 1 which compares IL-8 levels in patients with COPD with and without AAT deficiency, and in patients with bronchiectasis in the stable state and during an exacerbation.15 ,70 ,71
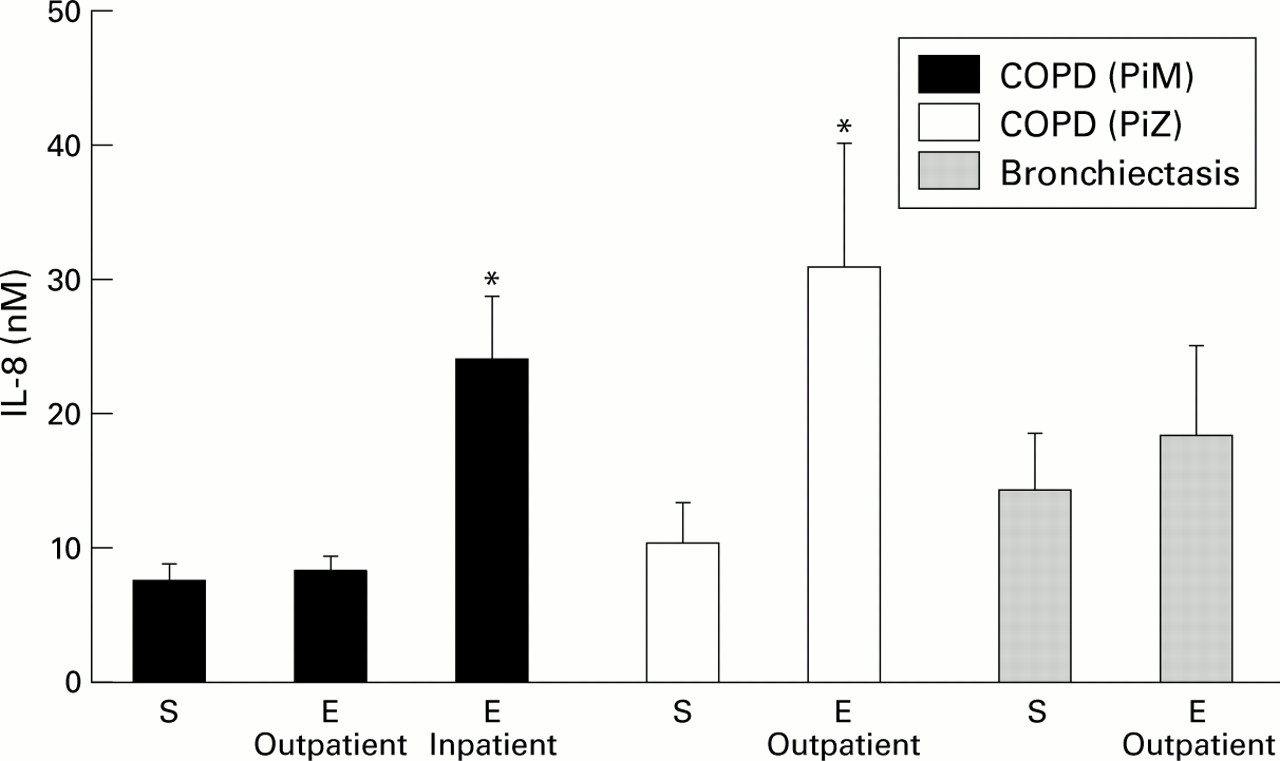
Concentrations of IL-8 (nM) in patients with COPD with (PiZ) and without (PiM) α1-antitrypsin deficiency, and in patients with bronchiectasis in the stable state (S) and during an exacerbation (E). Both inpatient and outpatient exacerbations are shown. The histograms represent mean (SE) values derived from Crooks et al,15 Hill et al,70 and Gompertz et al.71 *p<0.05 versus patients in the stable clinical state.
Technical factors
INDUCED SPUTUM
Not all patients with asthma or COPD regularly expectorate sputum in the stable clinical state. There has therefore been much interest in assessing airway inflammation using the technique of sputum induction in patients with either asthma or COPD, even when patients also expectorate spontaneously. There are several aspects that may influence the concentrations of airway inflammatory mediators including the process itself (whole sample versus selected plugs), the effect of the varying concentrations of hypertonic saline used (3–5%), the length of the induction process, the effects of dithiothreitol (DTT), and the effects of repetitive induction.
The effect of the concentration of hypertonic saline is not currently known and requires further study as it may potentially disrupt ionic binding of proteins and mediators. In addition, little is known about whether the length of sputum induction influences airways inflammation, although a study by Bhowmik et al found that sputum induction for seven or 14 minutes had no influence on sputum cell counts, cell viability, or cytokine measurement.72However, induction does influence airway inflammation and, after a 24 hour period, a repeated induction identifies increased neutrophil counts.73 ,74
The main methods of processing induced sputum involve either selection of sputum plugs or use of the entire sputum sample. This has been compared in healthy and asthmatic subjects and both methods have been found to have the same diagnostic value in distinguishing asthmatics from healthy subjects, although selected plugs had a higher proportion of eosinophils, a slight increase in viable non-squamous cells, and higher concentrations of ECP than the whole sample.75
Comparison of induced and spontaneous sputum samples in patients with COPD72 and asthma76 has demonstrated higher cell viability in induced samples, less squamous cell contamination, and better quality cytospins.76 Whether this relates to lysis of less viable cells as a result of hypertonic saline required to induce expectoration remains to be determined. Total and differential cell counts are similar in both types of sample but other measurements can be altered—for example, lower fibrinogen levels have been found in induced samples.76 Bayley et alfound that sputum induction (whole sample) produced approximately a 50% dilution in a number of inflammatory parameters including MPO, elastase activity, IL-8, LTB4, and albumin leakage77 compared with levels in spontaneous sputum from the same patients.
The authors routinely use and have validated assays to measure inflammatory mediators in sputum sol samples obtained by ultracentrifugation of untreated whole spontaneous sputum.78 Many other workers use the reducing agent DTT to hydrolyse disulphide bonds and to render sputum samples less viscous before processing and cell counting.61 ,72 ,76 However, DTT may have effects on the disulphide bonds within inflammatory molecules themselves (such as IL-8) or may alter their release from treated sputum macromolecules. Efthimiadis et al found that DTT affected both cell numbers and viability and increased the levels of ECP in induced sputum from asthmatic patients79 but had no effect on the levels of fibrinogen, IL-5, or IL-8. Other authors have found a significant reduction in EPO and MPO levels in DTT treated induced sputum from asthmatic patients but no effect on ECP.80 It is probably wisest to retain an aliquot of sample without DTT for cytokine assays unless the assays have been formally validated for samples treated with DTT.
Smoking
Prevalence studies in at least 10 countries have confirmed the association between smoking and the development of chronic bronchitis.81 Cigarette smoke recruits inflammatory cells to the lungs and studies have shown increased numbers of neutrophils in bronchoalveolar lavage fluid from smokers.82 ,83 In addition, the lungs of smokers with airflow obstruction contain more neutrophils than smokers without airflow obstruction84 ,85and the number of neutrophils recovered is related to the amount smoked.86
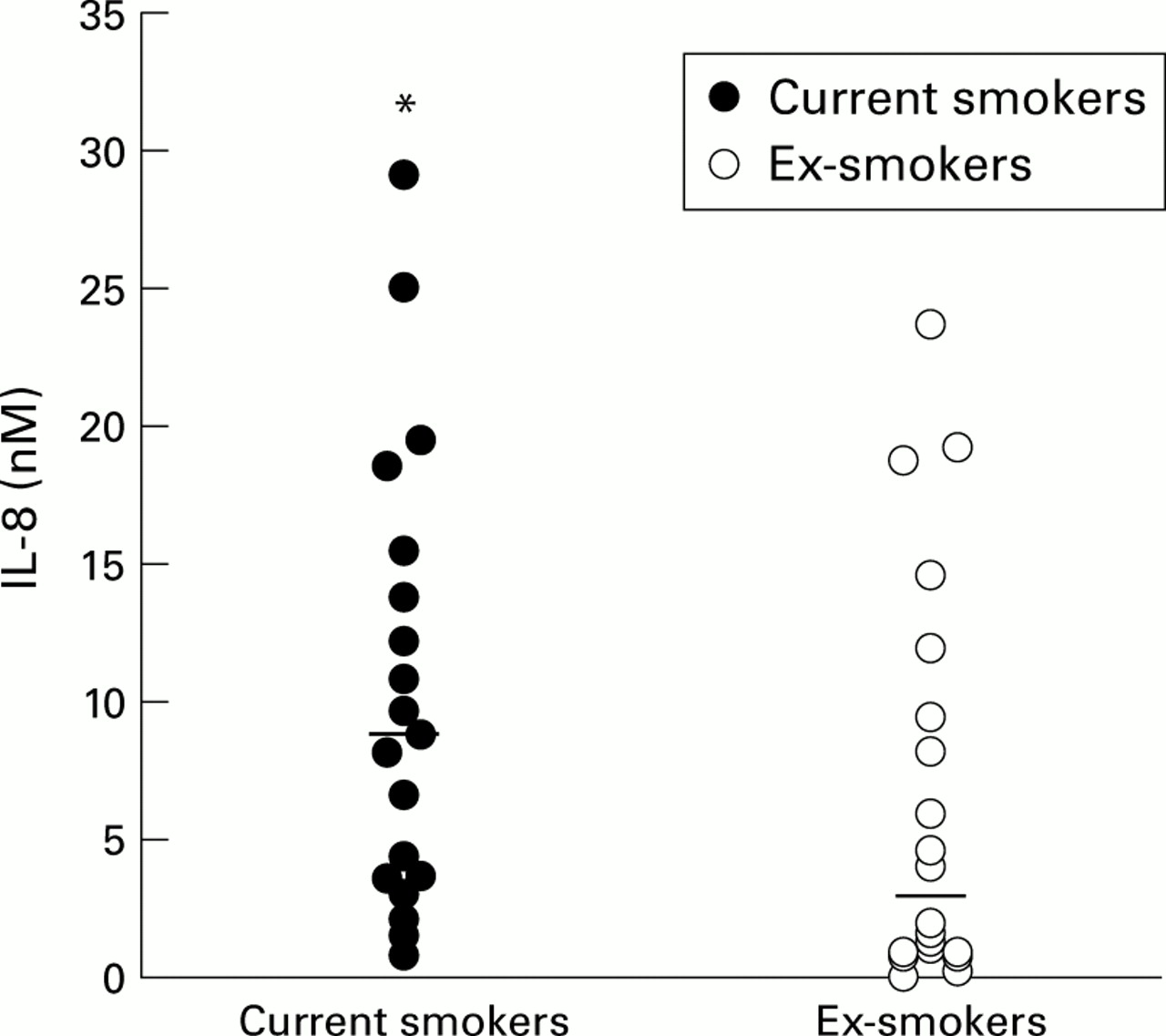
Continued smoking seems to be the major stimulus to airways inflammation in patients with chronic bronchitis and emphysema, whereas smoking cessation is believed to be beneficial in reducing the rate of lung function decline. In studies of patients with chronic obstructive bronchitis, current smokers had higher sputum (induced and spontaneous) IL-8 concentrations.20 ,87 One hypothesis is that cessation of smoking in patients with COPD leads to a reduction in airways IL-8 concentration (fig 2) which in turn reduces neutrophil recruitment, thereby providing a mechanism to explain the beneficial effect of smoking cessation on progression of neutrophil mediated lung disease.87 The reason for this effect is not clear at present but may relate to a reduction in epithelial production of IL-8 which can be induced by cigarette smoke.22
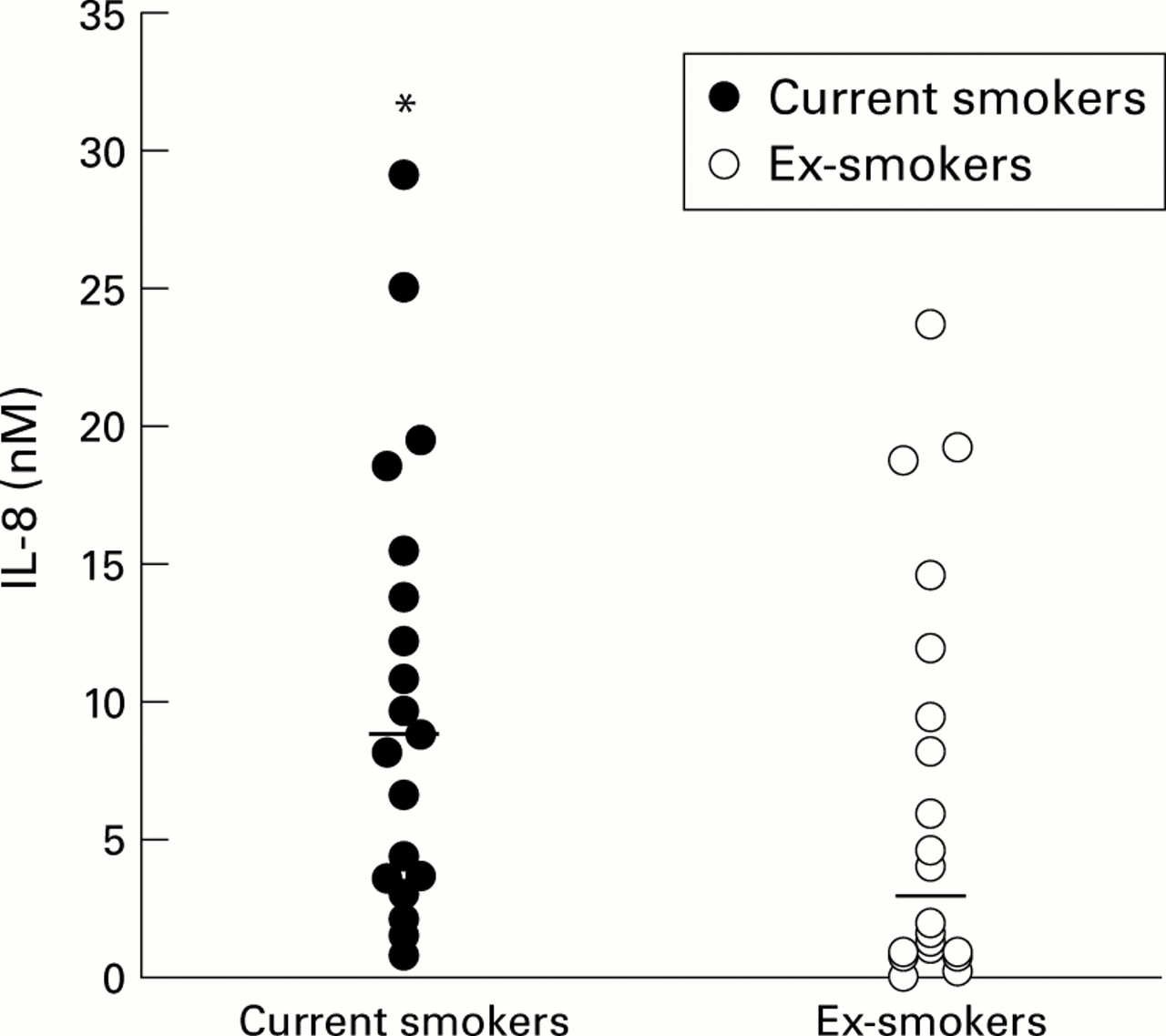
Individual IL-8 concentrations (nM) in current and ex-smokers. Median concentrations are indicated by the horizontal bars. *p<0.05 versus ex-smokers. Data derived from Hill et al.87
AAT deficiency
Subjects with severe AAT deficiency (PiZ phenotype) have decreased (about 15–20% normal) circulating and alveolar concentrations of AAT which facilitates the development of early onset and rapidly progressive emphysema.88 About 30–40% of these patients have chronic bronchitis,88 ,89 although the nature of the airway inflammation measured in spontaneous sputum has only recently been studied.
The pathogenesis of emphysema in AAT deficient patients is becoming clearer. Initial lavage studies revealed that PiZ patients have increased numbers of neutrophils in the lower airways, suggesting a greater potential elastase burden than in smokers with emphysema who have normal levels of AAT.90 In addition, patients with PiZ AAT deficiency have reduced concentrations of immunoreactive AAT in both sputum12 and lavage fluids.13 ,90 The pathogenic mechanism for the development of lung disease is thought to result from the low AAT concentrations in the presence of an increased neutrophil burden, leading to inadequate control of neutrophil elastase activity. In addition, extensive connective tissue destruction by migrating neutrophils may be amplified in AAT deficient patients as the Z type AAT has a slightly lower association rate constant for elastase.91 Finally, the low AAT levels result in an inability to control elastase activity in a wide area around the activated neutrophil resulting in even more connective tissue destruction.92 ,93
In the airways, however, SLPI is believed to be the major inhibitor of elastase.12 Studies with spontaneous sputum have shown that inflammation is increased in the larger airways of patients with chronic obstructive bronchitis and AAT deficiency.69 ,87Sputum concentrations of MPO and LTB4 are higher which suggests that LTB4 is the major chemoattractant responsible for the increased neutrophil migration, as suggested by previous studies using bronchoalveolar lavage.32 The source of the LTB4 is uncertain, although Hubbard et al 32 suggested that it was released from alveolar macrophages as a direct effect of uninhibited elastase. Sputum elastase activity is also increased in patients with AAT deficiency and could have the same effect in the larger airways as that seen in bronchoalveolar lavage, stimulating LTB4 production by airway macrophages.
The elastase activity detected in the larger airways in patients with AAT deficiency is probably due to the combined effect of lower sputum concentrations of both AAT and SLPI. Indeed, the low SLPI concentration may be a more important determinant of elastase activity in the larger airway where it is the major inhibitor. The mechanisms involved in disturbing the elastase/anti-elastase balance are likely to be complex in the larger airways. For instance, although elastase has been shown to decrease SLPI secretion from epithelial cells,56 it will also increase epithelial cell permeability, resulting in greater AAT leakage from plasma. In subjects with normal AAT levels these two effects may balance each other, retaining the anti-elastase screen. However, in patients with AAT deficiency the low serum and hence secretion AAT level may be less effective as SLPI falls, resulting in a continued deficient screen in the larger airways and hence uncontrolled elastase activity and local damage.
Bacteriology
In animal models low numbers of bacteria instilled into the airways are efficiently cleared by the primary host defence mechanisms alone.94 However, as increasing numbers of organisms are instilled, secondary host defences are activated including neutrophil migration.94 ,95 This is likely to reflect the release of pro-inflammatory mediators resulting in an acute inflammatory response with recruitment of neutrophils.95 ,96 Some patients with COPD have persistent bacterial colonisation and it is likely that this contributes to the morbidity of the disease. In particular, the chronic bacterial load may stimulate secondary host defences and lead directly to persistent airway inflammation even in the stable clinical state,97 ,98 although this has received little clinical attention.
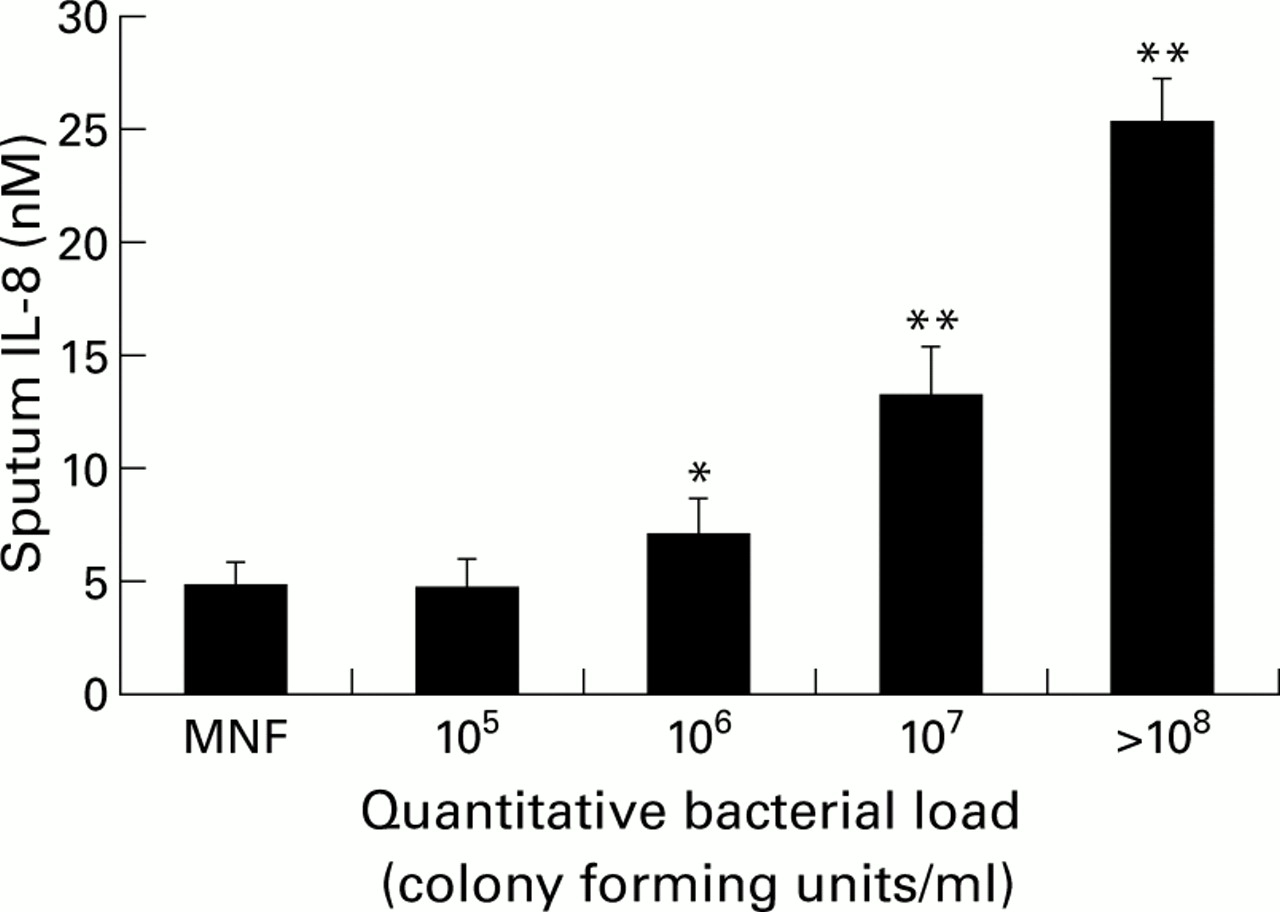
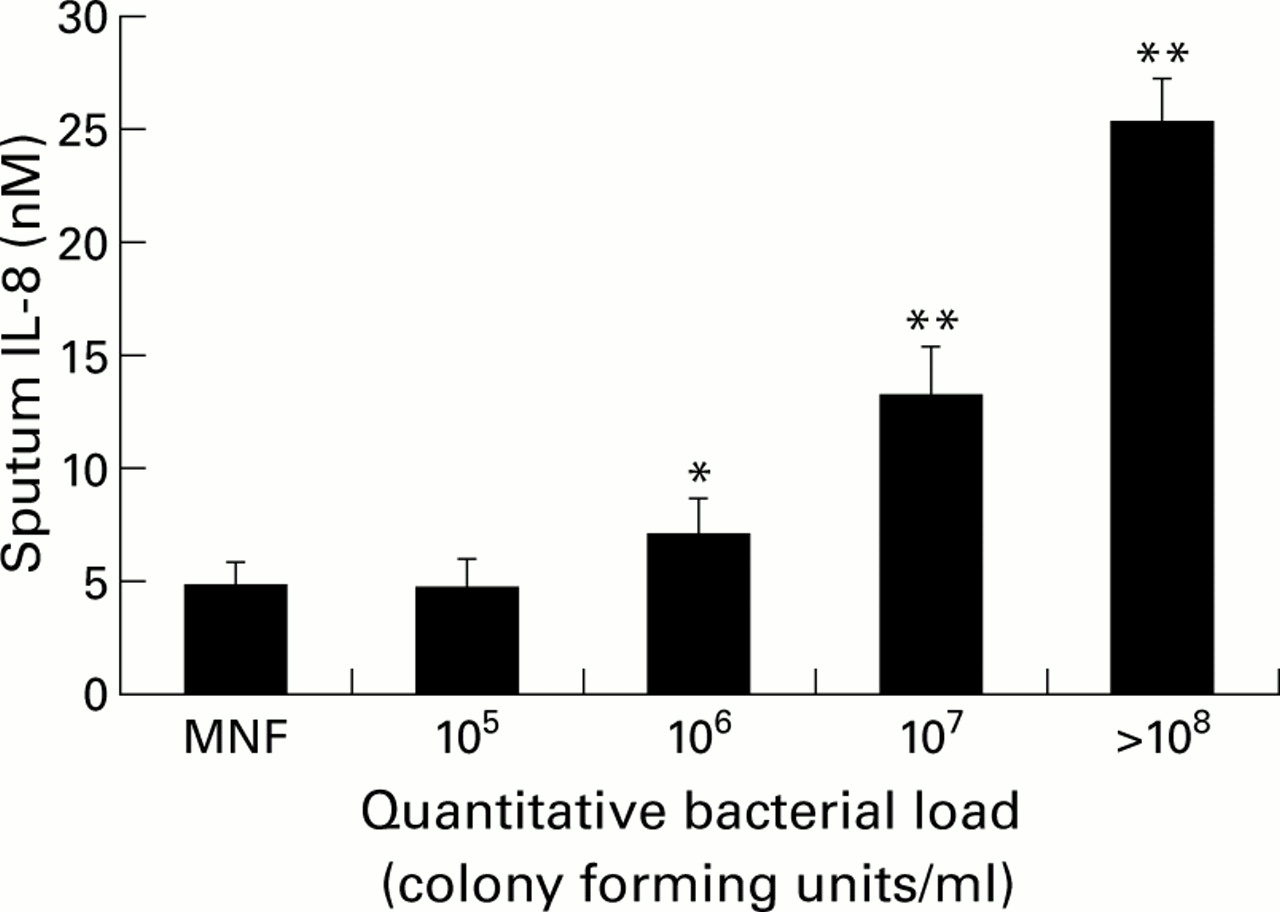
In a recent study by Hill et al a wide range of airway bacterial load was found among patients with chronic bronchitis while “clinically stable”. Increasing airway bacterial load was strongly correlated with the following in sputum: (1) increasing numbers of neutrophils (MPO); (2) increasing concentrations of the neutrophil chemoattractants IL-8 (fig 3) and LTB4; (3) increasing concentrations of active leucocyte elastase; and (4) increasing evidence of airway inflammation leading to albumin leakage.99 This has been supported by bronchoalveolar lavage data which showed that patients with stable COPD and potentially pathogenic organisms had a higher proportion of neutrophils and a higher TNF-α concentration.100 Thus, chronic bacterial colonisation can also influence airways inflammation significantly even when patients are “clinically stable” and needs to be taken into account when interpreting secretion data obtained from different patient groups.
{kind=link}
{kind=link}
{kind=link}
Relationship between sputum IL-8 concentrations (nM) and airway bacterial load (colony forming units/ml). MNF = mixed normal flora. Histograms represent mean (SE). Data derived from Hill et al.99 *p<0.05, **p<0.01 versus MNF.
Exacerbations
Airway inflammation would be expected to increase during exacerbations of COPD, in particular those that meet Anthonisen's criteria of increasing sputum volume and sputum purulence.101 Earlier work with spontaneous sputum found an increased proteinase burden during such exacerbations with increased protein leakage.58 Further work by Balbi and colleagues demonstrated a higher number of neutrophils and eosinophils in bronchoalveolar lavage specimens during exacerbations of chronic bronchitis,102 and bronchial biopsy specimens taken during such episodes have a marked eosinophilia and a milder increase in the number of neutrophils, activated CD3+ T lymphocytes, and TNF-α positive cells.103
A recent study by Hill et al used spontaneous sputum to investigate the nature of an exacerbation in patients with chronic bronchitis with and without AAT deficiency.70 The exacerbations filled all three Anthonisen criteria with increased sputum volume, purulence, and worsening breathlessness.101 At the start of the exacerbation there was increased airway inflammation reflected by a high sputum level of MPO, elastase activity, chemoattractant (LTB4), and evidence of protein leakage in patients with chronic obstructive bronchitis without AAT deficiency (table 1). Subjects with AAT deficiency also had increased airways inflammation at the start of their exacerbation and this was greater than in matched patients without AAT deficiency (table 1).70 Acute bacterial exacerbations in patients with chronic obstructive bronchitis that required hospital admission were associated with even greater airways inflammation (fig 1).15
Airway inflammation at the start of an exacerbation in outpatients with COPD, with and without severe α1-antitrypsin (AAT) deficiency
Treatment
The Isolde, Euroscop, and Copenhagen Lung studies have investigated whether inhaled corticosteroids reduce the rate of decline in forced expiratory volume in one second (FEV1) over a two year period in patients with COPD but have not directly measured airway inflammation. During two weeks of treatment with inhaled or oral corticosteroids Keatings et al could not demonstrate any effect on airway inflammation assessed with induced sputum.104 Similarly, Culpitt et al showed that inhaled steroids given over four weeks had no anti-inflammatory effect.105 However, two other longer studies have shown that treatment for eight weeks with inhaled steroids reduced chemotactic activity and increased elastase inhibitory capacity in spontaneous sputum106 and reduced the number of neutrophils in induced sputum.107 In addition, other investigators have shown that oral steroids can reduce eosinophils and ECP levels in patients with COPD with eosinophilia4 and that oral steroids reduced vascular protein leakage.108 On balance therefore it is likely that steroids do alter airway inflammation and again this should be borne in mind when assessing the results of patient studies.
Conclusion
A number of markers can be measured in sputum to assess airway inflammation. However, the process is complex in vivo and many factors including the disease itself, clinical state, current smoking history, colonisation, and treatment may influence the results. In addition, the processing of samples can influence the results and assay validation in the biological fluid therefore becomes critical. Many of the studies published in the literature involve small numbers of patients and many (if not all) of the factors have not been noted or stratified for. Future studies therefore need to bear this in mind and should ensure validation of all assays. The interpretation of the complex interaction between various mediators will probably only be clarified by the use of specific intervention strategies.