Article Text

Statistics from Altmetric.com
In 1951 the pathologists Churg and Strauss identified 13 patients who presented with a clinical syndrome characterised by asthma, hypereosinophilia, and evidence of vasculitis affecting a number of organs.1 The three main histological features found on pathological examination of these cases were extravascular granulomas, tissue eosinophilia, and necrotising vasculitis (fig 1). Most of these patients had been previously diagnosed with “periarteritis nodosa”. After reviewing a number of cases of periarteritis nodosa without asthma and finding no evidence of an eosinophilic granulomatous process, Churg and Strauss suggested that their 13 cases represented a separate disease process and coined the term “allergic granulomatosis and angiitis”. Later “Churg-Strauss syndrome” (CSS) became the accepted title of this distinctive form of systemic vasculitis. More recent pathological case series involving patients with CSS have highlighted the fact that not all patients have the three main histological features originally described by Churg and Strauss.2 ,3 Given the absence of a histological definition, it was proposed by Lanham et althat the following clinical criteria were required for the diagnosis of CSS: asthma, peripheral blood eosinophilia >1.5 × 109/l (or >10% of total white cell count), and evidence of a systemic vasculitis involving two or more extrapulmonary organs.2 More recently the American College of Rheumatology has developed diagnostic criteria for CSS when there is biopsy proven vasculitis.4 In this review we provide a clinical overview of CSS, review the evidence on which treatment protocols are based, and discuss the current understanding of the pathogenesis of the disease. In addition, we highlight recent controversies in the classification of systemic vasculitis and possible alternative management strategies.
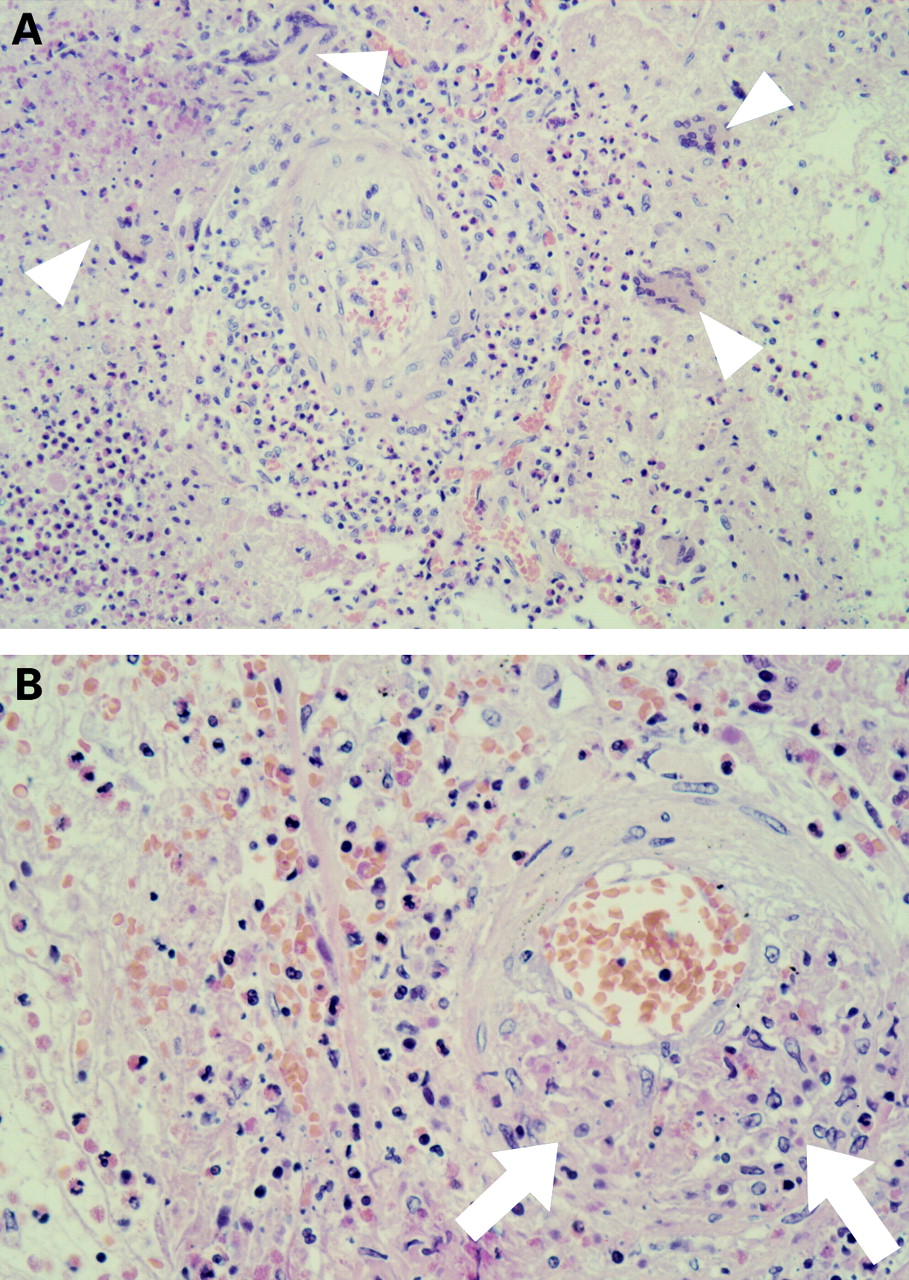
Open lung biopsy specimen from a patient with Churg-Strauss syndrome demonstrating the three main histological criteria originally described by Churg and Strauss. (A) Extravascular granulomas (arrowheads) are seen in association with an eosinophilic interstitial and alveolar infiltrate. (B) A higher power view highlights the necrotising vasculitic process (arrows).
Classification of vasculitis
Pulmonary vascular inflammation is seen most frequently as a manifestation of primary systemic vasculitis, but also occurs in association with a number of conditions including rheumatological disorders—for example, systemic lupus erythematosus and polymyositis, chronic infection, lymphoma, sarcoidosis, and extrinsic allergic alveolitis.5-10 Primary vasculitic processes affecting the lungs include giant cell arteritis, pulmonary capillaritis, Takayasu's arteritis, and those associated with circulating antibodies to neutrophil cytoplasmic enzymes (ANCA)—CSS, Wegener's granulomatosis (WG), and microscopic polyangiitis (MPA). Although definitive classification of systemic vasculitis in individual patients may not necessarily affect proposed management strategies, disease characterisation is important when designing clinical trials. By the early 1990s it became clear that a standardised system for the classification of systemic vasculitis was required to eliminate some of the controversy that had arisen from the comparison of clinical trials that had enrolled patients using different diagnostic criteria. As a result, in 1993 a consensus conference was held in Chapel Hill, USA where it was agreed that systemic vasculitis should be classified on the basis of vessel size (box FB1).11 Although the Chapel Hill vasculitis classification system emphasising vessel size is now generally well accepted, some discussion still surrounds the nomenclature of the forms of vasculitis that may involve medium sized vessels (CSS, WG, polyarteritis nodosa (PAN), and MPA) and the role that ANCA plays in their diagnosis. Although CSS is classified as a “small vessel vasculitis”, inflammation of medium sized vessels may occur. Similarly, a medium sized vasculitis can occur in MPA, but diagnosis requires inflammation of small vessels usually in the form of a segmental necrotising glomerulonephritis. The current definition of PAN includes only patients with an isolated medium vessel vasculitis.
Chapel Hill Conference proposal for the classification of primary vasculitis.11
Clinical features
The diagnosis of CSS should be suspected in patients presenting with hypereosinophilia, asthma, pulmonary infiltrates, and clinical evidence of vasculitis. Often patients have longstanding asthma and are taking systemic corticosteroids that can mask the symptoms of systemic vasculitis. CSS occurs with equal frequency in both sexes and can present at any age, with the mean age of onset being 40 years.2 ,3 ,12 ,13 It has been recognised that many patients progress through a prodrome of increasingly severe asthma to a stage characterised by eosinophilic pulmonary or gastrointestinal infiltration, before eventually developing vasculitis.2While this observation raises interesting questions regarding the pathogenesis of CSS, it does not assist in the diagnosis as the prodromal phase would describe many asthmatics and eosinophilic infiltration in the absence of vasculitis is a feature of all forms of eosinophilic pneumonia. Although respiratory symptoms are the most common presenting feature of CSS, the site of the vasculitic process is often outside the lungs, most commonly involving the peripheral nervous system, heart, skin, kidneys, and gastrointestinal tract. Evidence of vasculitis involving one of these extrapulmonary sites differentiates CSS from allergic bronchopulmonary aspergillosis (ABPA) and chronic eosinophilic pneumonia.
PULMONARY INVOLVEMENT
Asthma is the unifying feature of all patients with CSS and is frequently associated with allergic rhinitis (55–70%), nasal polyps, and sinusitis.2 ,3 In comparison to patients with common atopic asthma, asthmatic symptoms in CSS are said to develop at a later age and become more severe with time. In one study three quarters of the patients had received oral corticosteroids for asthmatic symptoms prior to the diagnosis of CSS.12 62–77% of patients with CSS have an abnormal chest radiograph on presentation.12 ,13 Infiltrates are often transient and can be associated with pleural effusions (29%).2 In a study investigating the spectrum of changes seen on computed tomography, Worthy et al reported that 87% of patients had radiological abnormalities, with a peripherally distributed parenchymal infiltrate being the most common finding (fig2).14 Unfortunately, this pattern of disease is not specific to CSS, being seen in other conditions such as chronic eosinophilic pneumonia and ABPA, which are also characterised by hypereosinophilia and an increased incidence of asthmatic symptoms. Other radiological abnormalities seen on computed tomography in this series included pleural and pericardial effusions, cavitating pulmonary nodules, bronchial wall thickening, and thickened interlobular septa.

{kind=link}
{kind=link}
Computed tomographic (CT) scan of a patient with Churg-Strauss syndrome showing a peripherally distributed parenchymal infiltrate that is also characteristic of eosinophilic pneumonia.
NEUROPATHY
Involvement of the neurological system is the most distinctive feature of the vasculitic process in CSS, with a peripheral neuropathy in the form of a mononeuritis multiplex occurring in up to 75% of patients.2 Central nervous system involvement is less frequent but has been described.15
MUSCULOCUTANEOUS
Cutaneous lesions in the form of vasculitic purpura, livedo reticularis, or subcutaneous nodules are seen in approximately two thirds of patients and reflect the propensity of CSS to involve small vessels. A migratory small joint arthritis occurs in 50% of patients.2
CARDIOVASCULAR INVOLVEMENT
Cardiovascular involvement is common in CSS and was the cause of death in 48% of the 50 cases reviewed by Lanham et al.2 Electrocardiographic (ECG) abnormalities are seen in 50% of cases and evidence of congestive cardiac failure caused by an eosinophilic myocarditis or coronary vessel vasculitis occurs in 25%.3 Pericardial effusions are also common and may cause constrictive symptoms.
RENAL LESIONS
A focal and segmental necrotising glomerulonephritis is the most common renal abnormality seen in CSS, occurring in 20–47% of patients.2 ,3 Other forms of renal disease also occur, including an eosinophilic tubular infiltrate, granulomas, and vasculitis. Renal failure is an uncommon complication of CSS, but has been described.16
GASTROINTESTINAL INVOLVEMENT
Abdominal pain in CSS is almost universal and may be caused by an eosinophilic gastroenteritis or vasculitis. The occurrence of pancreatitis, gastrointestinal perforation, or haemorrhage is more suggestive of an underlying vasculitis and, in a recently published study, these complications were found to be predictive of a poor outlook.17
Investigation and differential diagnosis
Broadly speaking, the investigation of a patient with suspected CSS involves exclusion of the known causes of eosinophilic pneumonia and establishing the presence of a vasculitic process. In 1936 Löeffler described the original cases of “pulmonary eosinophilia” in patients who developed recurrent lymphangitis, lymphatic obstruction, and fleeting pulmonary infiltrates in response to infection with the filarial species Wuchereria bancrofti. 18 Since that time the classification of eosinophilic pneumonia has been confused by the application of terms such as “Löeffler's syndrome”, “tropical eosinophilia”, and “pulmonary eosinophilia” to patients presenting with pulmonary infiltrates and hypereosinophilia. In a comprehensive review of eosinophilic lung disease by Liebow and Carrington it was felt that “eosinophilic pneumonia” would be the most appropriate term to be applied to this group of conditions (box FB2).19Investigation of the known causes of eosinophilic pneumonia requires exclusion of ABPA along with a detailed travel and drug history. Although parasitic infestation is most common following travel to endemic areas, domestic contact with dogs and immunosuppression are risk factors for Toxocara andStrongyloides infection, respectively, and requires consideration.
Classification of eosinophilic pneumonias
Once the known causes of eosinophilic pneumonia have been excluded, the clinical issue revolves around establishing the presence of vasculitis. A clinical presentation of asthma, hypereosinophilia, and mononeuritis multiplex is quite specific and was felt by Lanhamet al to be sufficient for a diagnosis of CSS.2 When the diagnosis is less typical, or when there is evidence of vasculitis involving organs from which tissue is more easily obtained, every effort should be made to obtain histological evidence of the vasculitic process before exposing the patient to potentially toxic therapy. A skin or renal biopsy is of proven value in systemic vasculitis and should be performed if cutaneous lesions or an active urinary sediment is present.20 Myocardial vasculitis is usually a clinical diagnosis based on the presence of signs of cardiac failure and supported by ECG or echocardiographic abnormalities. Non-specific ST segment and T wave changes are the most common ECG changes, while an echocardiogram generally shows global left ventricular dysfunction with a small or moderate sized pericardial effusion.
The clinical utility of ANCA in CSS raises the controversial issue of the sensitivity and specificity of ANCA in systemic vasculitis. These antibodies directed against proteins in the cytoplasm of neutrophils were first described in 1982 in a cohort of eight patients with glomerulonephritis, five of whom had coexistent pulmonary disease.21 Publications followed soon after highlighting their association with WG22 and later CSS.23The most commonly used method to detect ANCA involves indirect immunofluorescence staining of ethanol fixed neutrophils from healthy volunteers that have been incubated with the patient's serum. Three patterns of granulocyte staining can be observed: cytoplasmic (cANCA), perinuclear (pANCA), or atypical ANCA (any positive staining other than cANCA or pANCA). In CSS the prevalence of a positive serum ANCA result ranges from 44% to 66% with the most common pattern being perinuclear.17 ,24 ,25 A positive ANCA requires determination of antigenic specificity using an enzyme linked immunosorbent assay (ELISA). This is of particular importance with pANCA positive serum samples because this pattern of staining is often caused by antigens other than myeloperoxidase (MPO) and the clinical significance of a positive pANCA to a neutrophil cytoplasmic antigen other than MPO is uncertain. MPO is found to be the antigen causing the pANCA staining in only 50% of serum samples from patients with CSS,17 the remainder of the positive ANCA results being caused by a number of other neutrophil antigens (table 1). Our current understanding of the value of ANCA is also influenced by the expanding list of conditions that have been found to be associated with ANCA positive sera (table 2). Many of these conditions can present in similar ways to systemic vasculitis. Despite the limitations of ANCA as a screening test for systemic vasculitis, it is a useful investigation when combined with a positive ELISA to MPO when tissue from an affected organ is difficult to obtain. A pANCA pattern of staining in the absence of a positive ELISA is of low specificity for the detection of systemic vasculitis and should be ignored.45 Studies suggesting that a rising ANCA titre are predictive of relapse in WG led investigators to suggest that this investigation could be of value when tailoring immunosuppressive therapy.46 While it is true that changes in ANCA titre may reflect disease activity in a subgroup of patients with WG, recent studies do not support this approach in routine clinical practice.47 ,48 The relationship of ANCA titre to disease activity in CSS has not been looked at systematically but, based on the evidence from the literature on WG, ANCA should remain a diagnostic investigation only.
Target antigens in ANCA positive sera
ANCA in diseases other than systemic vasculitis
Although the clinical presentation of CSS is often characteristic, other forms of ANCA positive vasculitis may present with a similar pattern of organ involvement. For example, renal involvement is seen in most patients with ANCA associated vasculitis and sinus symptoms are present in 70% of patients with CSS and up to 92% of patients with WG.3 ,49 In an attempt to assist the differentiation of different forms of systemic vasculitis, the American College of Rheumatology devised the following criteria for the classification of CSS: asthma, eosinophilia >10% of total white cell count, mononeuropathy or polyneuropathy, migratory pulmonary infiltrates, paranasal sinus abnormality, and extravascular eosinophils.4 These criteria were based on a multivariate analysis of clinical, radiological, and histological variables obtained from 20 patients with CSS and 787 control patients with other forms of vasculitis. When there was histological evidence of necrotising vasculitis, the presence of four of six criteria was 85% sensitive and 99.7% specific for the diagnosis of CSS. It is important that these criteria are not used in the absence of histologically proven vasculitis as in this situation they are insensitive and poorly specific. For example, patients with either ABPA or chronic eosinophilic pneumonia can have asthma, sinusitis, pulmonary infiltrates, and a peripheral blood eosinophilia, fulfilling the criteria for the diagnosis of CSS.
Treatment
The value of corticosteroids in systemic vasculitis was established over 30 years ago. In 1967 Frohert et al reported that the five year survival of a cohort of patients with systemic vasculitis improved from less than 10% to 48% with the introduction of corticosteroids.50 Corticosteroids were found to be equally efficacious in a later study looking specifically at CSS.51 Since that time the French Vasculitis Study Group has enrolled patients with CSS and PAN in five consecutive studies aimed at assessing the value of various treatments that could be used as an adjunct to corticosteroids. In the first study patients received either prednisolone commencing at 1 mg/kg/day in combination with six months of plasma exchange, or the same regime with the addition of oral cyclophosphamide 2 mg/kg/day for 12 months. Overall there was no significant difference in the 10 year survival of the two groups (72% versus 75%), but patients receiving cyclophosphamide did have a reduced incidence of relapse and an improved clinical response to treatment.52 In the second trial patients received either prednisolone and plasma exchange or prednisolone alone.53 The addition of plasma exchange to prednisolone did not improve survival or reduce the rate of relapse. The elimination of plasma exchange as an effective treatment in the second trial led the investigators to conclude that reduced rate of relapse in the prednisolone, cyclophosphamide, and plasma exchange arm of the first trial was due to the cyclophosphamide. Unfortunately, the validity of this conclusion has not been confirmed by a trial comparing prednisolone and cyclophosphamide with prednisolone alone. The third and fourth trials involved the stratification of patients according to the presence of clinical features that had been previously established to be predictive of a poor prognosis: serum creatinine >140 μmol/l, proteinuria >1 g/day, gastointestinal involvement, cardiac failure, or central nervous system involvement.54 Patients with a good prognosis were assigned to receive either prednisolone and oral cyclophosphamide or prednisolone and intravenous cyclophosphamide (0.6 g/m2 monthly), while those patients in the poor prognostic group received prednisolone and intravenous cyclophosphamide alone or the same regime with plasma exchange. Based on the results of the third and fourth studies it was concluded that in patients with a good prognosis the two cyclophosphamide regimes were of equal efficacy, and the addition of plasma exchange was of no benefit to patients with clinical markers predictive of a poor prognosis.15 ,55Although these trials have been criticised for including patients with both PAN and CSS, a recently published retrospective follow up including only the CSS patients who had been enrolled in these studies largely supported the earlier prospective findings.17
The reduced rate of relapse provided by oral cyclophosphamide in CSS must be balanced against the increased risk of serious infection and urological malignancy associated with the use of this medication in systemic vasculitis. In an analysis of 207 patients receiving cyclophosphamide for vasculitis Bradley et al found that 10% experienced a serious infection and that most of the infections occurred soon after treatment commenced, in the absence of leukopenia and despite adequate drug monitoring.56 These data support the prophylactic use of trimethoprim 160 mg/sulphamethoxazole 800 mg three times weekly in patients receiving cyclophosphamide for systemic vasculitis, although the value of this practice has not been addressed prospectively. The incidence of haemorrhagic cystitis following treatment with oral cyclophosphamide for non-malignant conditions is 9–17%57 ,58 and it is estimated that the risk of urological malignancy is increased 15–45 times.59-62 The risk of developing haemorrhagic cystitis appears to increase with the cumulative dose of cyclophosphamide,63 although it has been reported in patients who have received as little as 100 mg.64 The bladder has the greatest exposure to acrolein, the urotoxic metabolite of cyclophosphamide, and is therefore the most frequent site of malignancy, although the cancer risk is increased at all sites along the urogenital tract. Strategies that reduce the cumulative dose of cyclophosphamide or use alternative immunosuppressive agents have been considered in an effort to reduce the risk of infection, haemorrhagic cystitis and, possibly, malignancy. Intravenous cyclophosphamide reduces the cumulative dose of the drug and was found to be of similar efficacy to oral cyclophosphamide in the third French Vasculitis Study Group trial.55Unfortunately, the finding that intravenous and oral cyclophosphamide was equally efficacious in CSS was based on small patient numbers and has not been supported by larger studies involving patients with other forms of systemic vasculitis. In WG the risk of relapse is increased with the use of intravenous compared with oral cyclophosphamide.65 ,66 The substitution of cyclophosphamide by mycophenolate mofetil or azathioprine after 4–6 months is another way of reducing the cumulative dose of cyclophosphamide and is standard practice in many centres. It also allows the reintroduction of cyclophosphamide at a later date, should relapse occur. Clinical experience suggests that abbreviating the course of cyclophosphamide does not increase the risk of relapse, but there is little evidence to support this practice. Trials aimed at determining the optimal duration of cyclophosphamide treatment are currently in progress.
The most appropriate way to screen patients for urothelial malignancies is unknown. Urinary cytology is an insensitive method of detecting urological malignancies but is often performed if patients develop haematuria.59 In one series 22% of patients with a history of cyclophosphamide induced haemorrhagic cystitis developed bladder carcinoma.57 Based on this finding, it was advocated that patients with a history of haemorrhagic cystitis should receive a follow up cystoscopy for further episodes of dysuria or haematuria, combined with six monthly urinary cytological tests and routine annual cystoscopy. In patients who develop haemorrhagic cystitis azathioprine is often substituted for cyclophosphamide, although non-randomised trials have suggested that azathioprine is not as effective as cyclophosphamide for the primary treatment of systemic vasculitis.61 ,62 Mycophenolate mofetil may be a more effective alternative to azathioprine in patients who have developed haemorrhagic cystitis as it has been reported to induce remission of other forms of systemic vasculitis that have been refractory to azathioprine treatment.67 A small number of patients with CSS fail to respond to prednisolone and cyclophosphamide. Anecdotal reports suggest that either antithymocyte globulin or pooled intravenous immunoglobulin are effective rescue therapies in this situation.68-71
Corticosteroid induced loss of bone mineral density is another treatment related side effect common in patients with systemic vasculitis. A follow up study of 96 patients with CSS found that 88% remained on long term oral prednisolone for asthmatic symptoms and a significant number developed corticosteroid induced osteoporosis.17 Current management guidelines for corticosteroid induced osteoporosis emphasise the value of excluding secondary causes of osteoporosis, which is of particular relevance to CSS as hypogonadism secondary to cyclophosphamide treatment is common.72 Current evidence suggests that vitamin D and calcium supplements are only partially effective in preventing corticosteroid induced osteoporosis and that bisphosphonates are the most effective primary and secondary preventative agents.73-75
Pathogenesis
Despite similarities in the histological pattern of vascular injury occurring in the various forms of systemic vasculitis, it has long been believed that these diseases are the result of different pathogenic mechanisms. This concept was suggested as early as 1954 when Godman and Churg proposed that CCS, WG and MPA (“polyangiitis” as it was then known) were a related group of vasculitidies likely to be caused by similar immune mechanisms.76 It was not until the development of immunofluorescence microscopy during the 1960s that scientific evidence emerged to support this theory. With immunofluorescence microscopy investigators were able to show that some forms of vasculitis were associated with the vascular deposition of antibody and complement. It was proposed that these forms of vasculitis were the result of immune complex deposition. The best described of this group of vasculitidies are essential cryoglobulinaemia and Henoch-Schonlein purpura characterised by the deposition of cryoglobulin and IgA antibodies, respectively. Initially, immune complex deposition was also favoured as the mechanism of vascular injury in CSS, although the majority of cases were “pauci-immune”, showing no evidence of immune protein deposition in the vascular wall. It was thought that microbiological antigens were triggering vascular injury in a situation analogous to hepatitis B associated vasculitis.77 This theory was supported by a retrospective study which reported that patients with CSS were more likely to have been vaccinated or to have received immunotherapy in the period immediately preceding the diagnosis.78 The failure of later studies to support these epidemiological associations and the paucity of detectable vascular immune deposits in the majority of patients meant that many investigators were reluctant to accept immune complex deposition as an explanation for the pathogenesis of CSS.
The discovery of ANCA and the recognition that these antibodies may contribute to disease pathogenesis was an important step in the development of a plausible disease model to explain the pathogenesis of CSS. Evidence implicating ANCA in vasculitis includes reports that neutrophils located in the walls of inflamed blood vessels have upregulated surface expression of proteinase 3 (PR-3) and MPO.79 ,80 Binding of ANCA to this surface expressed PR-3 and MPO can result in degranulation and enhanced superoxide production by neutrophils, which is a recognised cause of vascular injury.80 ,81 Vascular endothelial cells also have receptors for PR-3 and MPO, which could promote further vascular injury by marginating activated neutrophils onto the vascular wall.82 ANCA may also contribute to vascular inflammation by binding directly to endothelial cells. Binding of ANCA to the vascular endothelium leads to upregulation of IL-8 that is a potent inducer of neutrophil chemotaxis and could amplify any neutrophil mediated process causing vascular injury.83 Finally, ANCA also upregulates tissue factor production by endothelial cells which is a potent inducer of the alternative coagulation pathway and may further contribute to vascular injury.84 The stimuli for ANCA production and neutrophil activation in CSS are currently unknown but are the subject of ongoing investigation.
Despite the weight of evidence supporting the role of ANCA in the pathogenesis of CSS, recent case reports describing the development of CSS in patients receiving leukotriene receptor antagonists have forced investigators to revisit the possibility that foreign antigens may be important in the pathogenesis of the disease.85-88 Many of these case reports noted that CSS was diagnosed following reduction in the dose of corticosteroids, leading to the suggestion that the reduced dose of corticosteroids was the factor that precipitated the “unmasking” of the vasculitic process. This theory is supported by existing reports of asthmatic patients not receiving leukotriene receptor antagonists who developed CSS following weaning off oral corticosteroids.89 An alternative explanation is that CSS represents a hypersensitivity reaction to this class of medication. Drug induced vasculitis is rare but has been described in association with a number of other medications including carbamazepine, propylthiouracil, allopurinol, and quinine.90-93 Although the weaning off corticosteroids is an attractive explanation for the proposed association of leukotriene receptor antagonists with CSS, it is important to note that similar cases of CSS “unmasked” by reduction in corticosteroid dose were not reported when inhaled corticosteroids were introduced. Furthermore, there are now a number of reports of CSS developing in patients who have not received corticosteroids soon after the introduction of leukotriene receptor antagonists.94-96 Until there is a documented increase in the incidence of CSS or a plausible pathogenic model to explain the involvement of leukotriene receptor antagonists in vascular inflammation, any link between CSS and this class of drug remains speculative.
Conclusion
CSS is a disease characterised by asthma, hypereosinophilia, and vasculitis. Investigation of patients with suspected CSS requires exclusion of other causes of eosinophilic pneumonia and establishing the presence of vasculitis. Although a clinical approach to the diagnosis of CSS is appropriate when the presentation is classical, histological confirmation of vasculitis is reassuring as there is a significant risk of treatment related morbidity. In the absence of histological confirmation of a vasculitic process, a positive ANCA with antigenic specificity to MPO helps support the diagnosis of CSS. Evidence from a number of well conducted clinical trials supports the use of high dose corticosteroids and oral cyclophosphamide as first line treatment for CSS, although the addition of cyclophosphamide has not been shown to improve survival. Little is known of the pathogenic mechanisms that trigger CSS, but it is believed that ANCA contribute to the disease process. It is hoped that a greater understanding of the pathogenesis of CSS and other ANCA associated vasculitidies will lead to the development of more effective treatments specifically targeted at the immune mechanisms underlying these conditions.
Acknowledgments
The authors acknowledge Dr Andrew Nicholson, Department of Pathology, Royal Brompton Hospital and Professor David Hansell, Department of Radiology, Royal Brompton Hospital for their assistance in providing histological sections and radiological reproductions for use in this article.