Article Text

Statistics from Altmetric.com
Pulmonary alveolar proteinosis was first described by Rosenet al in 1958.1 It is an unusual diffuse lung disease characterised by the accumulation of large amounts of a phospholipoproteinaceous material in the alveoli. It has a variable clinical presentation and course. Most cases are primary but occasionally the condition is secondary to other conditions or inhalation of chemicals. Whole lung lavage remains the most effective treatment and the overall prognosis is good. Surfactant homeostasis is abnormal and animal experiments suggest that this may relate, in some instances at least, to defects in GM-CSF signalling. There are at least two congenital forms of the disease and several different animal models suggesting that pulmonary alveolar proteinosis is unlikely to be a single disease entity and more likely to represent a clinical syndrome.
Epidemiology
Pulmonary alveolar proteinosis is a rare lung disease and accurate estimates of incidence are not available. Current estimates suggest an incidence of one in two million people. The series reported in the literature suggest a male preponderance (male:female ratio 3:1).1-11 Peak onset is in the third or fourth decade of life with over 80% of reported cases occurring in this age group.1-9 However, there are reports of the disease occurring in neonates,12 children,13-15 and the elderly.11
Clinical features
CLINICAL PRESENTATION
Dyspnoea is the most common presenting symptom. It usually occurs on moderate exertion but in a few patients occurs at rest.1-11 Cough is the other common symptom. These symptoms are often trivial and some patients do not present until they develop a supervening infection. This may explain the acute onset of symptoms and fever observed in some patients. A low grade fever may also occur as a consequence of pulmonary alveolar proteinosis in the absence of secondary infection.6
Physical examination is often normal and inspiratory crackles on auscultation is the commonest abnormality.1-11 Clubbing of the fingernails is present in about one third of cases. Patients with advanced disease may have central and peripheral cyanosis.1-11 Raised serum levels of lactate dehydrogenase (LDH),16-18 tumour markers,19-22 mucin like glycoprotein (KL-6),23 and the surfactant proteins A24 ,25and D26 have been observed in pulmonary alveolar proteinosis.
PULMONARY FUNCTION TESTS
The predominant abnormality is a restrictive ventilatory defect8-10 with a reduction in lung volumes and diffusion capacity. Obstructive ventilatory changes are unusual but may be observed in smokers. Hypoxaemia is a prominent finding and patients with extensive disease have an increased alveolar/arterial pressure difference (P(A–a)o 2)8-10 which widens further on exercise.8 This is thought to be due to shunting of blood from right to left through an intact pulmonary capillary bed perfusing large numbers of poorly ventilated alveoli filled with lipoproteinaceeous material.8 Other contributory mechanisms may be septal oedema and, in rare cases, a degree of interstitial fibrosis has been reported.10 ,27 ,28
Pulmonary function tests can be used to assess disease severity, progression, and response to treatment. Arterial blood gas tensions, alveolar-arterial oxygen gradients, and change in P(A–a)o 2 gradient on exercise are better predictors of disease severity and functional impairment.8 ,10 Kariman et al 10 suggested that patients with an arterial partial pressure of oxygen (Pao 2) of greater than 9.3 kPa (70 mm Hg) or P(A–a)o 2gradient of less than 5.3 kPa (40 mm Hg) are more likely to improve spontaneously whilst those who do not meet these criteria are more likely to progress.
RADIOLOGY
Accumulation of phospholipoproteinaceous material in the alveoli results in a non-specific radiographic pattern of air space consolidation. There are no ancillary signs or specific features of the distribution of the consolidation to suggest the diagnosis of alveolar proteinosis on the basis of radiographic appearances alone.29-32 The consolidation is usually bilateral and patchy and in some patients is very extensive, despite relatively mild respiratory symptoms.29 ,30 In some series of alveolar proteinosis the patchy consolidation was perihilar (bat's wing pattern; fig 1A) in up to 50% of cases.1 ,6 ,11 However, the distribution of consolidation is highly variable and may be predominantly peripheral or basal (fig 1B).30 ,31 The abnormalities may be asymmetrical in up to 20% of cases11and isolated lobar involvement has been reported.33 ,34Other radiographic patterns that reflect air space filling include poorly defined acinar nodules and ground glass opacification, both of which usually occur in conjunction with more widespread dense consolidation.35 Air bronchograms within the consolidated lung are not usually a feature.
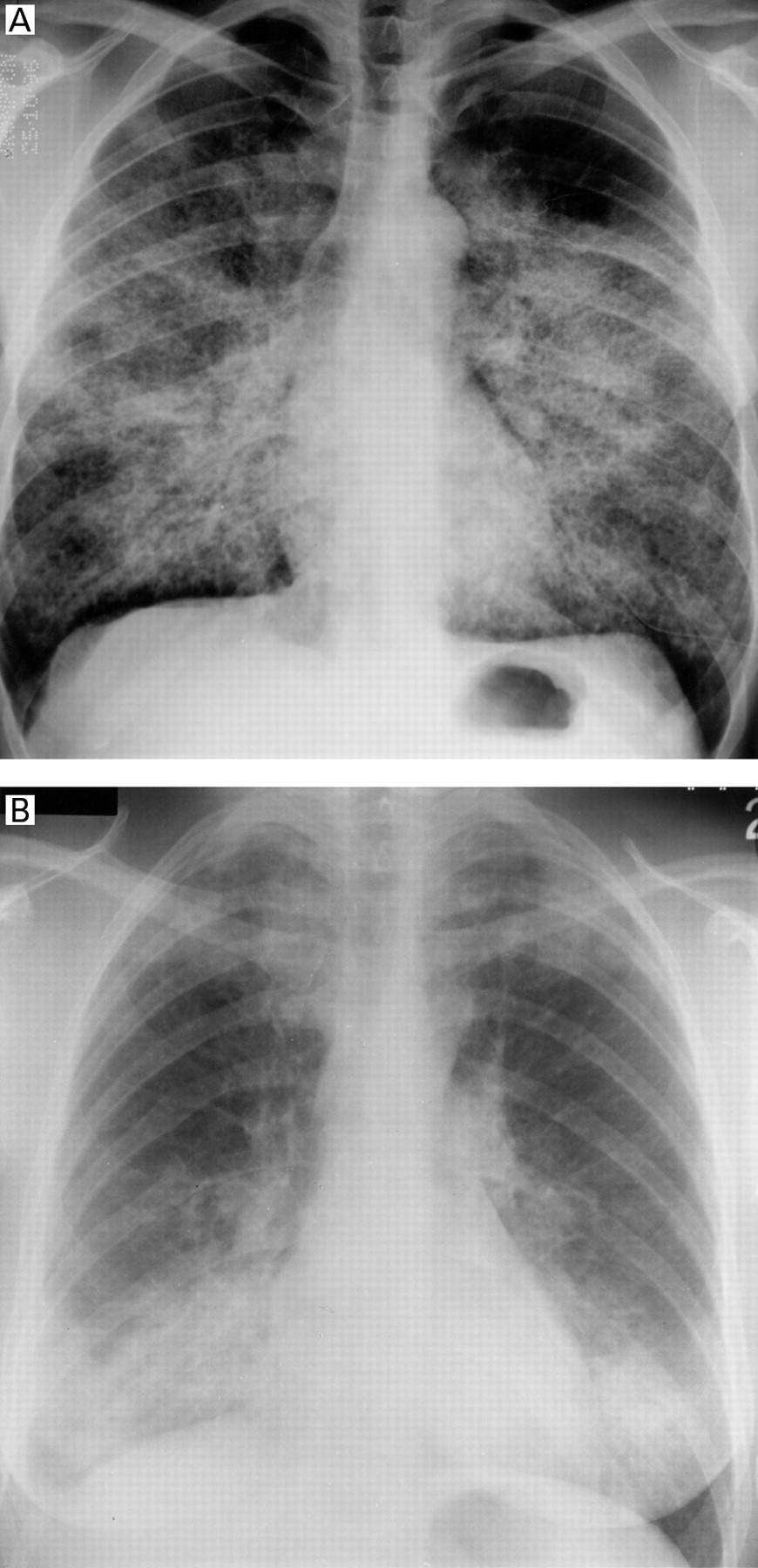
(A) Alveolar proteinosis: extensive confluent nodular shadowing which merges with consolidation in a predominantly perihilar distribution. (B) Unusual distribution of consolidation in a patient with alveolar proteinosis before lavage treatment. The shadowing is basal and apical with sparing of the mid zones.
High resolution computed tomographic (HRCT) scans of the thorax demonstrate the expected appearance of widespread air space consolidation, but also thickened interlobular septa, clearly visible within the affected lung and producing the so called “crazy paving” pattern (figs 2A and B).35 The extent of lung involvement is more easily defined on CT scans (which sometimes reveal sharply demarcated geographical areas of lung involvement). As with plain chest radiography, air bronchograms are not a feature despite widespread pulmonary consolidation. None of the CT studies published to date has identified a specific lobar or zonal distribution in this disease.36-38 Chest radiographs and HRCT scans of the thorax are of similar value for assessing disease severity.38 The appearances of alveolar proteinosis in children are not well characterised but a reticulonodular pattern, small nodules mimicking miliary disease, and a coalescence of various sized acinar nodules have been described.39

(A) Thin section CT scan through the upper lobes of a patient with alveolar proteinosis. Note the spared secondary pulmonary lobules in the posterior lung giving a geographical margin. Thickened interlobular septa are visible within the densely opacified parenchyma producing a “crazy paving” pattern. (B) Section through the lower lobes of another patient showing a similar pattern with several clearly demarcated patches of spared lung.
It was initially suggested that the crazy paving pattern in geographical areas of pulmonary opacification were specific for alveolar proteinosis. However, other conditions, most notably lipoid pneumonia40 and bronchioalveolar cell carcinoma,41 can sometimes mimic the CT features usually associated with alveolar proteinosis.
PATHOLOGY
The gross pathological findings in pulmonary alveolar proteinosis are patchy areas of yellow consolidation with an oily substance exuding from abraded surfaces. The classical finding on light microscopy is filling of the alveoli and terminal bronchioles with a granular lipoproteinaceous substance which stains a deep pink with periodic acid Schiff (PAS) stain.1 The alveolar architecture is usually well preserved although septal thickening from oedema37 or lymphocytic infiltration has been observed.42
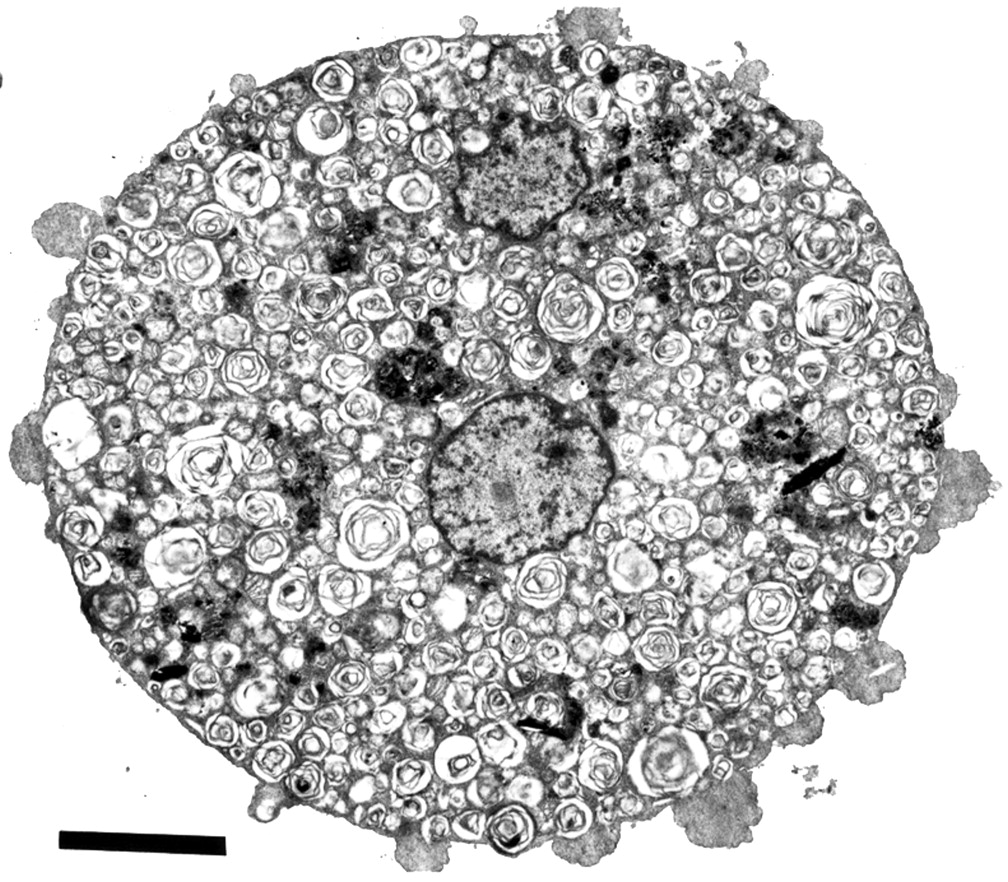
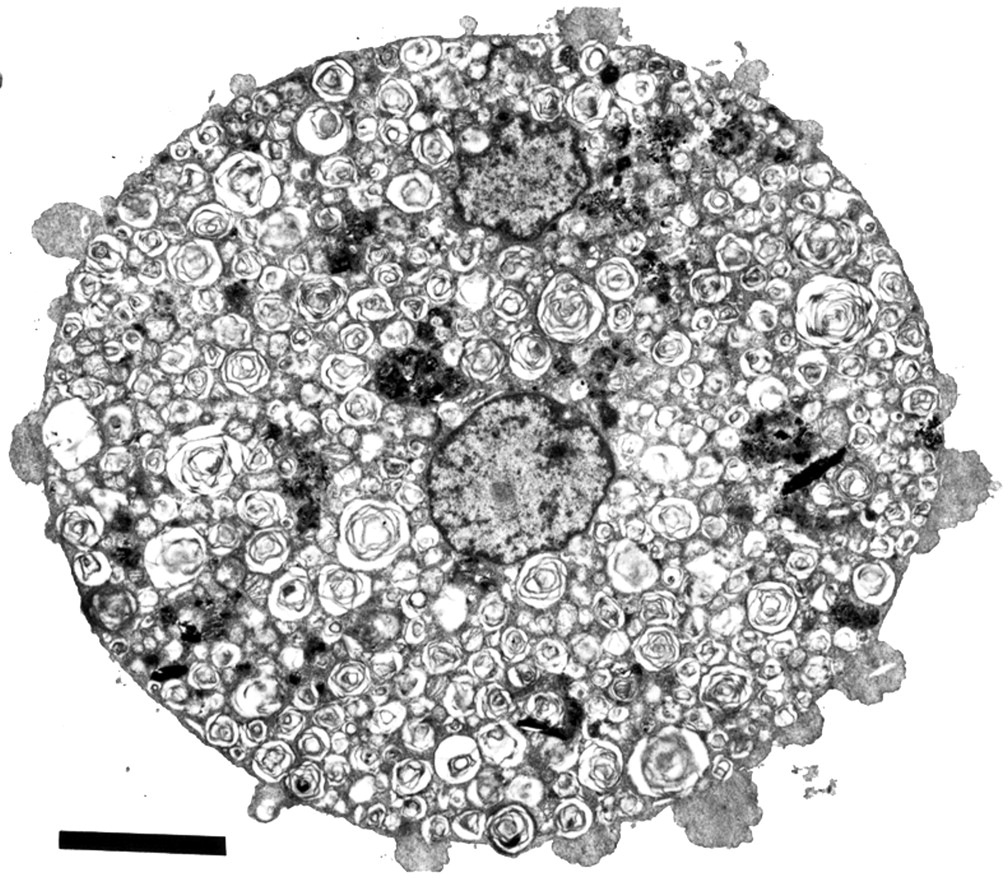
Ultrastructure analysis of lung specimens by transmission electron microscopy (TEM) shows abundant cellular debris and concentrically arranged laminated annular structures (lamellar bodies) within the alveolar lumen (fig 3).6 ,42-49 The structures range from uniformly spaced laminated structures (fig 4A) to irregular whorl-like structures with or without a dense osmiophilic core (fig4B).50 ,51 These structures represent phospholipids and are identical to inclusions found in normal type II pneumocytes.52 In contrast, tubular myelin structures are sparse (aggregates with well formed lattice structures that are formed in the alveoli under normal circumstances). The few alveolar macrophages that are present in the alveoli are enlarged and contain numerous complex phospholipoprotein inclusions (fig 5).53The inclusions are similar to the lamellar bodies and osmiophilic inclusions observed in the alveolar lumen and type II pneumocytes.

Electron micrograph of lung specimen showing cellular debris and lamellar bodies in the alveolar lumen. Bar represents 5 μm. (By courtesy of Ann Dewar, Electron Microscopy Unit, Royal Brompton Hospital and Imperial College of Science, Technology and Medicine at the National Heart & Lung Institute, London, UK).

Electron micrographs of lamellar bodies demonstrating (A) the uniformly arranged concentric laminated structure and (B) more irregular whorl-like structure with dense osmiophilic core. Bar represents 0.5 μm. (By courtesy of Ann Dewar, Electron Microscopy Unit, Royal Brompton Hospital and Imperial College of Science, Technology and Medicine at the National Heart & Lung Institute, London, UK).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electron micrograph of a foamy macrophage containing numerous complex phospholipid inclusions. Bar represents 5 μm. (By courtesy of Ann Dewar, Electron Microscopy Unit, Royal Brompton Hospital and Imperial College of Science, Technology and Medicine at the National Heart & Lung Institute, London, UK).
BRONCHOALVEOLAR LAVAGE
Milky fluid is usually obtained on bronchoalveolar lavage of an affected segment.54-56 Examination of the cytospin demonstrates a basophilic granular extracellular deposit with a few enlarged foamy macrophages and cellular debris on staining with May-Grünwald Giemsa. The extracellular substance is stained pink with PAS but negative with alcan blue, which allows the differentiation of the phospholipoprotein aggregates from mucins. The cellular content is usually sparse and differential cell counts of the lavage fluid is usually unhelpful in making the diagnosis; an inflammatory infiltrate may indicate a superimposed infection.57 ,58 The ultrastructural appearance on electron microscopy is characteristic with abundant lamellar bodies and cellular debris but only a few tubular myelin aggregates.50 ,54-56 ,59 ,60 The alveolar macrophages are usually enlarged with abundant lipid inclusions with the same ultrastructural characteristics as those found in the extracellular material.
Numerous studies have characterised the intra-alveolar material obtained following bronchoalveolar lavage in patients with pulmonary alveolar proteinosis. The major constituent of the lavage fluid is phospholipid, mainly lecithin, the main component of surfactant.61-63 The lavage material also contains serum proteins and surfactant specific proteins64-75 including increased concentration of surfactant protein A (SP-A)76and surfactant protein D.26 One of the SP-A oligomers in pulmonary alveolar proteinosis tends to form large aggregates which retain an ability to bind with phospholipids but fails to form the uniform lattice structure of tubular myelin.77
Various tumour markers have been detected in the bronchoalveolar lavage fluid of patients with pulmonary alveolar proteinosis.20 ,21 However, their diagnostic value is limited as they are found in other pulmonary diseases.78-80
DIAGNOSIS
The diagnosis of pulmonary alveolar proteinosis can be made with confidence on the basis of the appearance of the lung on the HRCT scan of the thorax35 ,38 in conjunction with an examination of lavage fluid obtained from segmental alveolar lavage.50 ,81 ,82 Examination by light microscopy is adequate83 but ultrastructural analysis by electron microscopy strengthens the diagnosis. Transbronchial biopsy specimens provide a tissue diagnosis84 but, in view of the patchy nature of the disease, may be inconclusive. The procedure also carries the risk of a pneumothorax or haemorrhage. Open lung biopsy specimens obtained by the video assisted thoracoscopic route have the same disadvantages, in addition to the potential for surgical complications. Bronchoalveolar lavage samples a larger distal lung field and is a much safer procedure.
Treatment of pulmonary alveolar proteinosis
Whole lung lavage is the safest and most effective form of treatment for alveolar proteinosis. Historically, treatment has included corticosteroids,6 potassium iodide,3and streptokinase5 with variable success. Ambroxol stimulates the intracellular formation and secretion of surfactant85 ,86 and, although this would be expected to exacerbate the intra-alveolar accumulation of surfactant, there is a single report of improvement in one patient following treatment.87 Aerosolised trypsin has been used to treat pulmonary alveolar proteinosis88-90 on the grounds that trypsin would hydrolyse the proteinaceous material and hence improve clearance. Some of the patients treated with trypsin developed an allergic reaction and there is the potential for proteolytic damage. However, none of these studies was controlled and the response observed in some patients may represent the spontaneous improvement that is seen in up to 25% of cases.1 ,6 More recent developments such as treatment with GM-CSF are discussed later.
WHOLE LUNG LAVAGE
Whole lung lavage is now considered the treatment of choice. The technique has evolved since the early instillation of heparinised saline via an intratracheal catheter.91 ,92 Although there are no randomised controlled studies, there is good evidence of efficacy with several studies showing improvement in exercise tolerance and symptoms following whole lung lavage8-10 ,50 whilst others have shown objective improvements in pulmonary function,9 ,10 arterial oxygenation,8 ,11 ,50and shunt fraction.8 ,10 Some studies have shown improvement following the lavage followed by a gradual decline and subsequent improvement following a repeat lavage.8 ,10Radiological clearing is less impressive immediately after lavage but occurs more gradually,93 the time period varying between individuals.93 ,94 Whole lung lavage is associated with improvements in macrophage function95 and a decreased incidence of opportunistic infections.8 ,10 ,94
INDICATIONS FOR WHOLE LUNG LAVAGE
The main indication for whole lung lavage is limitation in daily activities due to dyspnoea. The threshold for lavage is therefore lower for a person who works as a heavy labourer than someone who has a sedentary lifestyle. Some authors have suggested that patients with a Pao 2 of less than 9.3 kPa (70 mm Hg) or a P(A–a)o 2 of more than 5.3 kPa (40 mm Hg) are more likely to progress and hence require whole lung lavage.10
TECHNICAL ASPECTS OF WHOLE LUNG LAVAGE
The technique used at the Royal Brompton Hospital is described in detail and is similar to that practised at other centres. Following induction of general anaesthesia, an experienced anaesthetist introduces a double lumen endobronchial tube. A careful check is made to ensure that the two lungs are isolated. Both lungs are ventilated with 100% oxygen for at least 20 minutes to eliminate nitrogen from the alveolar gas. The lung to be treated is then isolated at the end of expiration. The volume of that lung at the end of expiration is estimated from preoperative measurements of functional residual capacity (FRC). The volume of oxygen is gradually absorbed from the non-ventilated lung at a rate of approximately 100 ml/min. Warm (36–37°C) neutral sterile saline (0.9% saline with 0.6 mmol sodium bicarbonate per litre saline) is instilled into the lung through a closed system at the same rate as oxygen is adsorbed until the estimated FRC value has been reached. At this point the lung is completely de-gassed and full of saline. Tidal volume increments (500–1200 ml) of the saline are let into the lung under gentle gravitational force—that is, a saline column of not more than 50 cm—and then let out again via the closed system into a measuring cylinder. The temperature, volume of saline instilled, and fluid balance is carefully controlled. The initial returns are typically very milky or turbid and the process of filling and draining the lungs with saline is repeated until the effluent becomes clear. Total volumes of saline required can range from 20 to 40 l. At the end of the procedure the residual saline is drained and aspirated from the lung and ventilation with 100% oxygen is resumed. The double lumen tube is replaced by an endotracheal tube and the patient transferred to the recovery unit for approximately one hour of postoperative ventilation. At the end of this recovery time any residual neuromuscular block is antagonised and the patient is awakened and extubated. Patients are transferred back to the ward within the following hour.
It has recently been possible to perform bilateral sequential whole lung lavage in the same treatment session. The principles are identical except that at the end of the lung lavage of the first lung a decision is made on the ability of the patient to tolerate one lung ventilation of the lung that has just been treated. The second lung is then isolated and treated as described above.
The process of whole lung lavage is slow (2–4 h) and requires an experienced theatre team and postoperative facilities. The key components to ensuring safety is an anaesthetist who is very skilful in the placement of the double lumen endobronchial tube and meticulous monitoring to ensure that the two lungs remain isolated throughout the procedure.
Some centres use manual percussion and positional drainage during lavage to improve the removal of surfactant. A randomised study showed that optical density of recovered lavage fluid was greater from patients who received manual percussion than in those who received percussion using mechanical devices or no percussion.96
A recent report suggested that manual ventilation during the second half of the drainage cycle might aid surfactant clearance,97 but this was a single report of an individual who failed to respond to conventional lavage. After half the instilled volume had drained, the lung was ventilated manually 20 times with a tidal volume of 500 ml and then allowed to drain freely. The clearance of surfactant was increased despite a considerable reduction in the volume of lavage fluid used.97 The technique requires controlled assessment as there is a greater potential for complications such as barotrauma and increased absorption of lavage fluid.
OUTCOME OF WHOLE LUNG LAVAGE
In our experience over 60% of patients have a good response within two lung washes per lung, a good response being defined as complete return to normality in terms of exertional capacity as judged by the patient. This is consistent with reports from the literature, which confirms that only a few patients require more than six cycles of lung lavage.8 ,10 ,11 ,94 A small proportion (<15%) require lavage every six months to maintain their functional status8 and fewer than 10% are non-responders.
SAFETY OF WHOLE LUNG LAVAGE
The major adverse effect from whole lung lavage is hypoxaemia which can be improved by ventilation with a high inspired oxygen concentration.91 ,98 ,99 Arterial oxygenation improves during the filling phase due to the increase in airway pressure and shunting of blood to the contralateral ventilated lung.99Emptying of the lung causes a decrease in airway pressure and perfusion of the surfactant filled alveoli creates a shunt in the lung undergoing treatment and hence a fall in Pao 2. 99 Haemodynamic changes occur with single lung ventilation98 ,100 but invasive monitoring is unnecessary in most cases.101 Hyperbaric oxygen,102 partial cardiopulmonary bypass,103and temporary venovenous extracorporeal gas exchange104 ,105 have been used as supportive measures for patients with severe hypoxaemia and in children undergoing whole lung lavage.
The major risks of whole lung lavage concern the correct placement of the double lumen endobronchial tube with overspill of lavage fluid into the ventilated lung being the main risk. Expert placement and checking for leaks prior to lavage and throughout the procedure are essential safety measures. Barotrauma can occur with rapid instillation of large volumes of fluid. Other reported complications include pleural collections, hydropneumothoraces, and surgical emphysema.10 ,11 The risk of hypothermia is minimised by careful monitoring of the patient's core temperature. The patient lies on a heated mattress and is covered with a convective heated blanket. The lavage fluid temperature is controlled using an in-line heat exchanger.
MULTIPLE SEGMENTAL LAVAGE
Multiple segmental or lobar lavage by fibreoptic bronchoscopy is a possible alternative to whole lung lavage.106 ,107 The advantages are that general anaesthesia is avoided and it may be performed in patients in whom whole lung lavage would be hazardous. However, the yield by this method is small and the volumes of lavage fluid limited. Hence, multiple lavages are required and this makes the procedure less tolerable than whole lung lavage. Lobar lavage with trypsin has also been attempted but the data are limited to two patients with no clear evidence of efficacy.107 This technique also carries the potential of proteolytic damage.
Prognosis
The major complication of pulmonary alveolar proteinosis is infection with unusual organisms such asAspergillus species,108 Nocardia species,109-113 Mycobacteriumspecies,17 ,114-117 Cryptococcus neoformans,118 Histoplasma capsulatum,119 Pneumocystis carinii,120 and viruses.121 This susceptibility is multifactorial; impaired macrophage function,122 impaired host defence due to abnormalities of surfactant proteins, and the rich intra-alveolar accumulations may favour the growth of micro-organisms.123 These unusual infections were responsible for some of the mortality associated with pulmonary alveolar proteinosis in the past.1 ,6 ,124 In contrast, we have had only one case of opportunistic infection complicating pulmonary alveolar proteinosis at our centre, an experience shared by other centres since the advent of treatment with whole lung lavage.8 ,10 ,94 Corticosteroids should not be used as empirical treatment for alveolar proteinosis due to its potential to exacerbate opportunistic infections.
The development of interstitial fibrosis in patients with pulmonary alveolar proteinosis has been reported in isolated cases,27 ,28 ,125-127 though whether this is a causal finding or coincidental occurrence is uncertain. One patient died from progressive pulmonary fibrosis.27 Other rare reports include the development of emphysematous bullae,127 ,128pneumothorax,129 and asphyxia due to alveolar flooding in a patient during general anaesthesia.130
The overall prognosis for alveolar proteinosis treated by whole lung lavage is excellent. There are also several reports of spontaneous resolution,131-134 and series prior to the introduction of whole lung lavage quote spontaneous improvement in up to 25% of patients.1 ,3 ,6
Pathogenesis
NORMAL SURFACTANT HOMEOSTASIS
Surfactant homeostasis is a complex dynamic process involving alveolar type II cells and macrophages. The hydrophobic (surfactant proteins B and C) and hydrophilic (surfactant proteins A and D) proteins associated with the surfactant and the phospholipids are synthesised and secreted mainly by alveolar type II cells. The importance of surfactant proteins B and C (SP-B and SP-C) in surfactant homeostasis is well established from findings such as their ability to accelerate the adsorption of lipids to an air-liquid interface.135 ,136 The roles of SP-A and SP-D on lipid interactions is less clear.
The freshly synthesised surfactant complex is packaged in lamellar bodies within alveolar type II cells and then released from these intracellular storage granules into the liquid hypophase covering the alveolar epithelium.137 The material secreted from the lamellar bodies is transformed by an as yet ill-defined pathway into a lattice like structure known as tubular myelin.138 From this structure the phospholipids spread over the epithelial surface to form a monolayer film, rich in dipalmitoylphosphatidylcholine, which reduces the surface tension forces tending to collapse the lung. Alveolar type II cells and alveolar macrophages have an important role in uptake, degradation, and recycling of surfactant.139-141 Surfactant protein A may play a part in the re-uptake of phospholipids and regulate the secretion of surfactant140-143 but its precise role remains unclear. The lipid monolayer also regulates secretion of phospholipid, presumably by altering exocytosis.144 This process is influenced by the amount of saturated phospholipid in the lipid monolayer.144
MACROPHAGE FUNCTION IN PULMONARY ALVEOLAR PROTEINOSIS
Alveolar macrophages in pulmonary alveolar proteinosis have defective chemotaxis,122phagocytosis,145 ,146 and phagolysosomal fusion.147 These defects are thought to be acquired since normal macrophages develop impaired phagocytic activity following incubation with lavage fluid from patients with pulmonary alveolar proteinosis. The lipoproteinaceous material also acts as a scavenger for free radicals and hence impairs the oxidative burst that occurs in macrophages during phagocytosis.148 Binding of SP-A and SP-D to the collectin receptor on alveolar macrophages initiates the oxidative burst in macrophages149 and enhances phagocytosis.150 Surfactant proteins bind to various bacteria,151 influenza A virus,152 andCryptococcus neoformans 153 and this facilitates the uptake of these organisms by alveolar macrophages. The important role of surfactant proteins in host defence may explain the higher incidence of opportunistic infections associated with pulmonary alveolar proteinosis.1 ,6
DISEASE ASSOCIATIONS
A number of cases of pulmonary alveolar proteinosis have been described in association with other diseases or a definite aetiological agent and have been termed secondary pulmonary alveolar proteinosis. Secondary pulmonary alveolar proteinosis can be distinguished from primary or idiopathic disease on the basis of the staining pattern of surfactant specific apoproteins in histological specimens.64 In primary cases a uniform staining pattern is seen while in the secondary form of the disease a patchy focal pattern occurs.64 Pulmonary alveolar proteinosis has been described in association with immunodeficiency states154 ,155 and haematological malignancies.124 It is more common in myeloid leukaemias156-161 but has also been reported in lymphomas,124 Fanconi's anaemia,162 ,163 and IgG monoclonal gammopathy.164 The overall incidence of secondary pulmonary alveolar proteinosis in patients with haematological malignancies is 5.3%, though it is higher in neutropenic patients (8.8%) and in those with acute myeloid leukaemia (10%).165 However, most cases are mild and require no intervention. Treatment of the underlying condition may produce improvement but whole lung lavage may need to be considered in patients who are very symptomatic.
In a number of cases of pulmonary alveolar proteinosis there has been a history of exposure to chemicals and mineral dusts.6 One study reported greater amounts of birefringent particles in lung tissue in over three quarters of patients with pulmonary alveolar proteinosis in comparison with controls.166 Specific reports have included exposure to aluminium dust,167titanium,168 and drugs such as busulfan and chlorambucil.169 Similar changes have been reported in sandblasters exposed to high levels of silica dust.170-172
Animal experiments have reproduced the pathological features of pulmonary alveolar proteinosis following exposure to a variety of mineral dusts and drugs such as iprindole.173-178However, extrapolating the experimental model to the human disease is difficult, especially as effects of agents are specific to species and dependent on dose and environmental conditions.43
GENETIC PREDISPOSITION
Congenital alveolar proteinosis was identified as an autosomal recessive inherited deficiency179 and a deficiency in SP-B was subsequently identified as the cause of the disease.180 Further studies have revealed two mutations in the SP-B gene, a frameshift mutation consisting of a substitution of three bases (GAA) for the single nucleotide C in codon 121 (121ins2)181 and a deletion of one base pair at (122delT),182 resulting in a premature stop codon and the absence of SP-B in surfactant. Congenital alveolar proteinosis is a genetically heterogenous disease exhibiting a wide range of phenotypic variations183 ,184 which implicates the involvement of other loci or novel SP-B mutations in its manifestations.183 Studies on cells cultured from patients with congenital alveolar proteinosis with normal SP-B protein levels have found normal levels of granulocyte colony stimulating factor (GM-CSF) but reduced expression of the βc receptor for GM-CSF/IL-3/IL-5 in three patients.185 In one of the patients a point mutation (proline to threonine at codon 602) in the βc receptor gene was identified.185
ANIMAL MODELS
Three murine models have shed new light on the mechanisms underlying alveolar proteinosis and strengthening the association of deficiencies in haemopoietic cells with alveolar proteinosis. Knockout mice of both GM-CSF186 ,187 and the GM-CSF receptor (generated by disruption of the gene for the murine common β chain of the GM-CSF/IL-3/IL-5 receptor)188 ,189 show perturbed surfactant homeostasis leading to pulmonary alveolar proteinosis. There is defective expression of the receptor on both alveolar macrophages and type II pneumocytes. Detailed studies of surfactant metabolism, incorporation of phospholipids, and expression of surfactant protein messenger RNA in these models suggest that the abnormalities of pulmonary alveolar proteinosis are likely to be due to abnormal surfactant catabolism rather than synthesis.190 ,191 The evidence also suggests that surfactant clearance is affected by defects in GM-CSF signalling.192 Defective GM-CSF receptor expression has been observed on leucocytes in acute myeloid leukaemia and may account for the high incidence of pulmonary alveolar proteinosis in this condition.193 The fourth gene implicated in alveolar preoteinosis is at theItch locus; it encodes a gene product of the ubiquitin machinery which is thought to influence the regulation of haemopoietic cell growth and differentiation. Disruption of this locus leads to a complex fatal disease which includes pulmonary inflammation and alveolar proteinosis.194
HYPOTHESIS OF DISEASE MECHANISMS IN PULMONARY ALVEOLAR PROTEINOSIS
The complex process of surfactant homeostasis, the occurrence of at least two different congenital forms, and the presence of different animal models suggest that pulmonary alveolar proteinosis represents a clinical syndrome rather than a single disease.
Deficiency or complete lack of SP-A is unlikely to be implicated in the pathogenesis of pulmonary alveolar proteinosis as illustrated by the murine models,195-197 but structural abnormalities of surfactant proteins may cause an imbalance in the surfactant life cycle. Surfactant proteins have a complex structure with distinct binding domains, and structural abnormalities that allow them to interact but not activate receptors may interfere with surfactant homeostasis. Abnormal oligomers of SP-A have been identified in patients with pulmonary alveolar proteinosis.198-200Abnormal multimerised SP-A isolated from patients with pulmonary alveolar proteinosis failed to form tubular myelin and had a lower affinity to bind to type II cells.199 The multimerised oligomer was a less effective inhibitor of lipid secretion.199 It is unclear whether the abnormal proteins are a consequence of alveolar stasis or the primary defect. The complex protein structure may be susceptible to denaturation and may explain the association of some cases of pulmonary alveolar proteinosis with exposure to high levels of inhaled minerals and chemicals.6
The presence of an antagonist for the surfactant protein receptors would be expected to have much the same effect. The removal of abnormal oligomers of surfactant proteins or receptor antagonists may explain the gradual improvements observed after lung lavage. Differential changes in surfactant composition leading to a greater decrease in SP-A following lung lavage also support this hypothesis.201 The various surfactant components have differing clearance rates and changes in composition following lavage suggests a restoration of normal surfactant homeostasis.
Evidence from the murine models and the occurrence of pulmonary alveolar proteinosis in haematological malignancy suggests that imbalances in cytokines, particularly IL-10 and GM-CSF, may lead to abnormal surfactant metabolism. Investigation of GM-CSF expression in one patient with pulmonary alveolar proteinosis suggested that expression of messenger RNA for GM-CSF was normal but there was a failure to secrete GM-CSF.202 This study demonstrated high basal levels of IL-10, a potent inhibitor of cytokine secretion at the transcription level,203 ,204 and the addition of IL-10 antibody normalised secretion of GM-CSF.202 These results are beginning to illustrate the complex role of cytokines in surfactant homeostasis. Secretagogues also regulate the number of cellular receptors205 acting through protein kinase C or cyclic AMP pathways to raise cytosolic Ca2+ levels. Abnormalities in intracellular processing and expression of receptors may also lead to abnormal surfactant homeostasis.
In murine models bone marrow transplantation reverses the intra-alveolar accumulation of surfactant206 but the lymphocytic infiltrate persists.207 Lung transplantation has been considered for some individuals with alveolar proteinosis but there is a report of recurrence following double lung transplantation.208 This suggests that a systemic factor may be involved in some cases of the disease. It is possible that there is a defect in cellular mechanisms or receptor involving macrophages and type II pneumocytes. Correcting only one of these defects by lung transplantation or a bone marrow transplant may not lead to a complete cure. Furthermore, the spontaneous occurrence of alveolar proteinosis has been reported in patients following lung transplantation.209
Future potential treatments
The murine models of GM-CSF deficiency emphasised the role of GM-CSF in surfactant homeostasis.187-190 One patient who failed to respond to lung lavage has been treated with recombinant GM-CSF with an improvement in exercise tolerance, partial clearing of alveolar infiltration on the chest radiograph, and an improvement in the P(A–a)o 2 gradient.210 There was reasonable evidence of efficacy in this single case with a time course pattern of improvement in the P(A–a)o 2gradient following treatment with GM-CSF, deterioration on discontinuation of treatment, and improvement on resumption of treatment. The treatment of three further patients with GM-CSF has been recently reported in abstract form.211 Only one patient responded and the degree of improvement was similar to previous improvements seen with whole lung lavage in that individual. Potential side effects of GM-CSF are local reactions, fever, myalgia, headache, and flu-like reactions. More serious side effects include anaphylaxis, cardiac failure, and leaky capillary syndrome. Other disadvantages are the need for a daily subcutaneous injection and the high cost of GM-CSF.
Gene therapy may be a future treatment option in patients with specific genetic defects. This may include patients with a congenital abnormality of surfactant protein B181-183 and defect in the β chain of the common receptor for GM-CSF/IL-3/IL-5.186 Other genetic disorders are likely to exist as illustrated by the gene knockout murine models. Transfection with DNA for SP-B and SP-A has been shown to express surfactant associated protein cDNA in vitro in human epithelial cell lines and in vivo in rats.212 The selective expression of GM-CSF in type II pneumocytes in GM-CSF deficient mice corrects the alveolar proteinosis observed in the deficient animals and highlights the potential of genetic therapy in the future.192
Summary
Pulmonary alveolar proteinosis represents a syndrome with a number of possible aetiologies. The appearances on the HRCT scan of the thorax will often suggest the diagnosis and should be confirmed by examination of bronchoalveolar lavage fluid. Whole lung lavage appears to be the most effective form of treatment and is a safe technique in experienced hands. The overall prognosis is excellent with treatment. A few patients are resistant to whole lung lavage. It is important to investigate these non-responders further as they may respond to some of the more targeted treatments.
Acknowledgments
The authors thank Ann Dewar for providing the electron micrographs used in this publication.