Article Text

Abstract
BACKGROUND Sequestration with associated cystic adenomatoid malformation is rare. A study was undertaken to determine whether pulmonary sequestration associated with congenital cystic adenomatoid malformation has a more favourable natural history than that of sequestration without associated cystic adenomatoid malformation.
METHODS An outline of the postnatal work up leading to the management of extralobar or intralobar pulmonary sequestration with congenital cystic adenomatoid malformation diagnosed antenatally as pulmonary malformation is presented and the indications for surgical intervention are discussed.
RESULTS In five infants in whom an antenatal ultrasound scan had detected a congenital lung malformation at 18–19 weeks gestation a final diagnosis of extralobar or intralobar pulmonary sequestration with congenital cystic adenomatoid malformation was made postnatally. Postnatal ultrasound and computerised axial tomographic scans confirmed the diagnosis of sequestration by delineating anomalous vascular supply. Cystic changes were also observed in the basal area of the sequestration in all patients. Four children remained asymptomatic and one infant presented at 10 months of age with pneumonia. The mean age at surgical resection was 6.8 months (range 2–10). Histopathological examination confirmed intralobar pulmonary sequestration with associated Stocker type 2 congenital cystic adenomatoid malformation in two patients and extralobar pulmonary sequestration with associated Stocker type 2 congenital cystic adenomatoid malformation in three patients. The mean period of follow up was four years (range 1–8). The children remain well and are developing normally.
CONCLUSIONS The importance of seeking an anomalous blood supply in children with congenital lung lesions is emphasised. Pulmonary sequestration and congenital cystic adenomatoid malformation probably share a common embryogenesis despite diverse morphology. The natural history of antenatally diagnosed lung masses is variable. Early postnatal surgical resection of pulmonary sequestration with cystic adenomatoid malformation is recommended. Surgical excision should be conservative, sparing the normal lung parenchyma.
- intralobar pulmonary sequestration
- extralobar pulmonary sequestration
- congenital cystic adenomatoid malformation
Statistics from Altmetric.com
- intralobar pulmonary sequestration
- extralobar pulmonary sequestration
- congenital cystic adenomatoid malformation
Pulmonary sequestrations are regions of lung parenchyma which lack a normal connection to the tracheobronchial tree and possess an anomalous systemic blood supply, usually from the aorta or its major branches. Sequestration has an estimated incidence of 0.15% to 1.7% in the general population1 and was first described by Rokitansky and Rektorzik in 1861.2 ,3 Pryce in 1946 introduced the term “sequestration” to describe the synonyms accessory pulmonary lobe, pulmonary aberration, supernumerary lung, nebenlunge, and Rokitansky lobe.4 Pulmonary sequestration is classified as intralobar, in which the abnormal tissue is contained within the normal lung, and extralobar, in which the sequestration is separate from the normal pulmonary lobe and outside the visceral pleura. Pulmonary sequestration is associated with many combinations of abnormalities and other concomitant anomalies such as congenital cystic adenomatoid malformation. Congenital cystic adenomatoid malformation is a focal pulmonary dysplasia, rather than a hamartoma, due to the presence of skeletal muscle in the cyst walls in most cases. It represents 25% of all congenital lung malformations and was first described by Ch’in and Tang in 1949.5 The occurrence of congenital cystic adenomatoid malformation within a sequestrated lobe is rare, with 17 cases of intralobar pulmonary sequestration with congenital cystic adenomatoid malformation and 19 cases of extralobar pulmonary sequestration with congenital cystic adenomatoid malformation having been described since 1949.6-19 The routine use of antenatal ultrasound has permitted early visualisation of fetal abnormalities and sequential scanning provides the opportunity to observe these anomalies to delivery. The aim of this paper is to review the literature and discuss the embryology, natural history, and the probable indicators for surgical intervention of pulmonary sequestration with congenital cystic adenomatoid malformation. An outline of the postnatal work up leading to the management of extralobar/intralobar pulmonary sequestration with congenital cystic adenomatoid malformation is also presented.
Methods
Results
In all five patients prenatal scanning diagnosed congenital lung abnormalities at 18 or 19 weeks of gestation (table 1). Sequential scanning showed that the evolution of the cysts remained uncomplicated during fetal life (in all patients) and the neonates were delivered at a mean gestation age of 38.2 weeks (range 33–40) with a mean birth weight of 2879 g (range 1544–3880) (table 2).
The mean age at surgical resection of the sequestration with associated basal cystic changes was 6.8 months (range 2–10). Histopathological examination confirmed intralobar pulmonary sequestration with associated Stocker type 2 congenital cystic adenomatoid malformation20 in two patients and extralobar pulmonary sequestration with associated Stocker type 2 congenital cystic adenomatoid malformation in three patients (tables 1 and 2). Table 3 summarises the characteristics of congenital cystic adenomatoid malformation as classified by Stocker.20 The mean period of follow up has been 4 years (range 1–8). The children remain well and are developing normally.
Congenital cystic adenomatoid malformation
INTRALOBAR PULMONARY SEQUESTRATION/CONGENITAL CYSTIC ADENOMATOID MALFORMATION (N = 2)
Patients 1 and 2
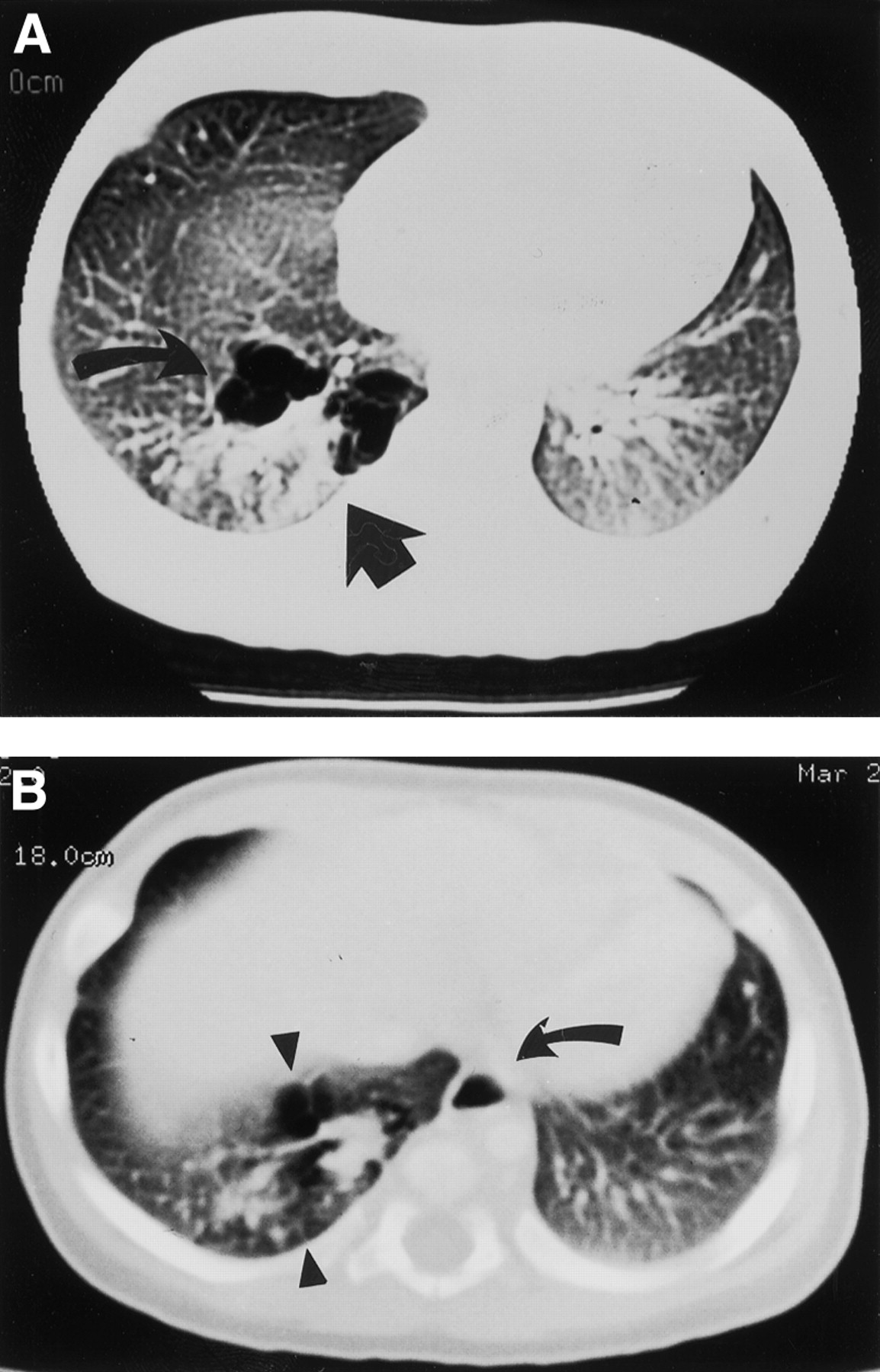
Computed axial tomographic (CT) scanning demonstrated a complex mass in the posteromedial aspect of the right lower lobe in patient 1 which consisted of multiple air-filled cysts of varying sizes and a solid component apparently contiguous with normal lung parenchyma. Two anomalous arteries, one arising from the abdominal aorta and passing superiorly and another arising low from the thoracic aorta, were delineated supplying the abnormal area of the lung (fig 1A and B). The venous drainage was not shown but a prominent hemiazygos vein branching low of the azygos was seen. In patient 2 the left lower lobe showed a similar complex mass with associated diffuse cystic changes. Two large aberrant systemic arteries arising from the abdominal aorta were delineated on the CT scan. The appearance in both patients was suggestive of intralobar pulmonary sequestration. The infants remained asymptomatic. An elective thoracotomy performed at a mean age of 4.5 months (range 2–7) revealed a lower lobe intralobar pulmonary sequestration with multiple cysts in the basal and medial segment of the sequestration. A right and a left lower lobectomy were performed respectively in both children (fig 2). The abnormal large systemic arterial blood supply to the intralobar pulmonary sequestration was from the paravertebral abdominal aorta traversing the inferior pulmonary ligament through the diaphragm, and another from the thoracic aorta in patient 1. Two large aberrant arteries arising from the abdominal aorta were identified in patient 2. The venous drainage was by four tortuous veins to the pulmonary vein (fig 2) in patient 1, and patient 2 had a normal venous drainage. The resected right lower lobe was 100 × 70 × 42 mm and the left lower lobe was 124 × 92 × 60 mm in size, each covered by a visceral pleura, and the cut surfaces showed multiple pale cystic areas. Histological examination showed that the cysts were lined by ciliated cuboidal to columnar epithelium and supported by attenuated smooth muscle (fig 3). Respiratory bronchioles and normal alveoli were present between the cysts. Elsewhere the alveolar development was normal with focal intra-alveolar haemorrhage and pulmonary oedema. There was no evidence of inflammation. The large vessels showed thick elastic laminae confirming systemic artery configuration. The diagnosis of intralobar pulmonary sequestration with associated congenital cystic adenomatoid malformation (Stocker type 2) was confirmed (fig3).

(A) CT scan of patient 1 showing a complex mass posteromedially in the right lobe comprising multiple air filled cysts of varying size (arrows) and a vessel arising from the aorta (arrow) and passing superiorly to supply the abnormal lung. (B) A further vessel lying left of the vertebral column behind the aorta (arrow) arising from the lower part of the thoracic aorta.

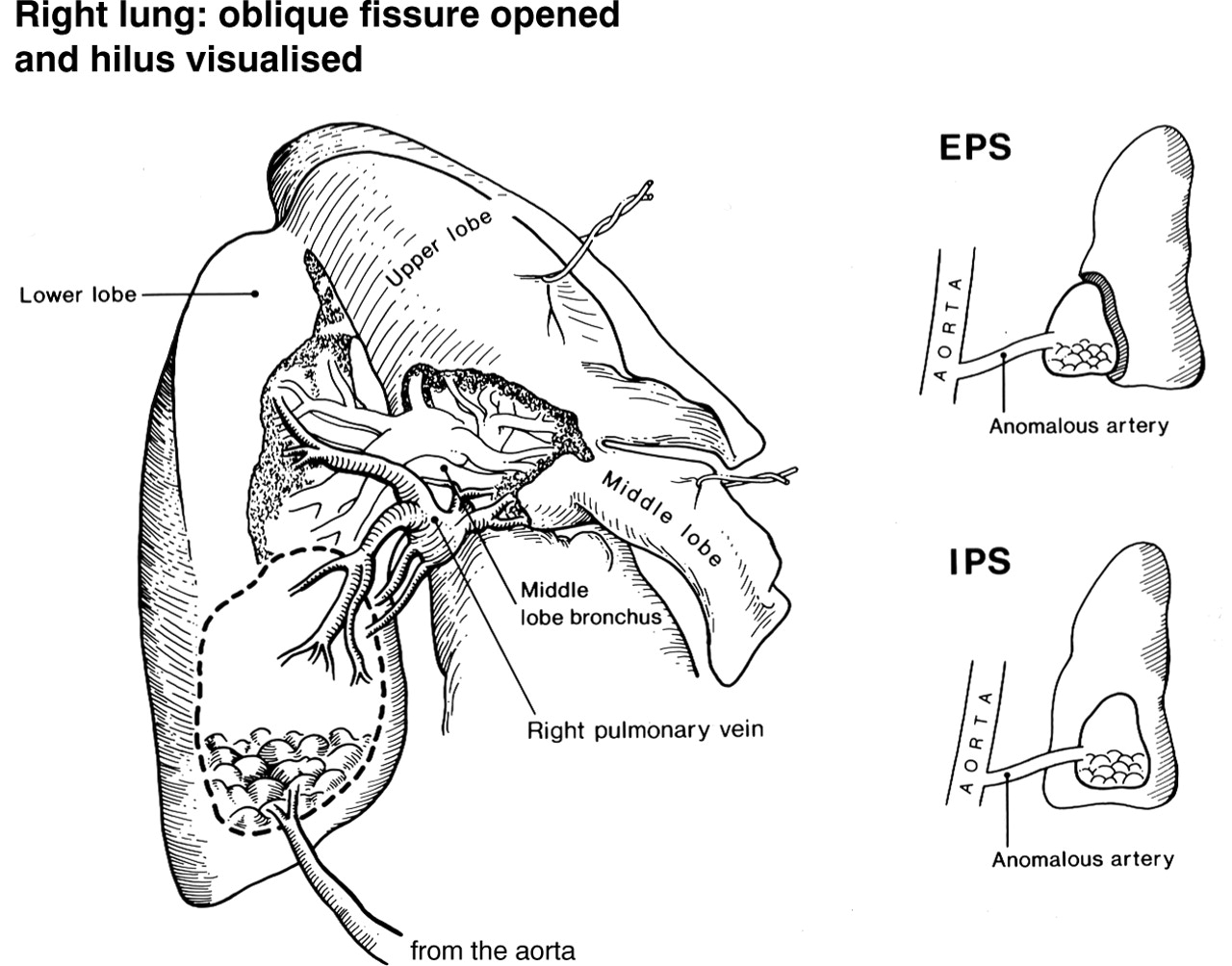
Diagram of intralobar pulmonary sequestration as seen at surgery. Large anomalous artery supplying the basal area of the intralobar pulmonary sequestration and four efferent veins draining into the pulmonary vein. Insert showing the difference between intralobar pulmonary sequestration (IPS) and extralobar pulmonary sequestration (EPS).

{kind=link}
{kind=link}
{kind=link}
(A) Low power microscopic view showing multiple irregulars cystic spaces interspersed with normal respiratory bronchioles and alveoli (B and C). The cystic spaces are back-to-back with the epithelium thrown up into papillary projections. The high power views demonstrate cysts lined by ciliated cuboidal to columnar epithelium and supported by attenuated smooth muscle (arrows). Simple cuboidal epithelium is also seen (arrows). Respiratory bronchioles and normal alveoli are present between the cysts and normal non-dilated terminal bronchioles, communicating with normal alveoli, with focal intra-alveolar haemorrhage. There is no evidence of inflammation. The histological features are of Stocker type 2 congenital cystic adenomatoid malformation within a sequestrated lobe due to the presence of normal distal lung parenchyma.
EXTRALOBAR PULMONARY SEQUESTRATION/CONGENITAL CYSTIC ADENOMATOID MALFORMATION (N = 3)
Patients 3, 4, and 5
The mean age at thoracotomy was 8.3 months (range 6–10). Thoracotomy revealed extralobar pulmonary sequestration with multiple cysts in the posterior basal area of the sequestrated lobe. The anomalous arterial supply to the sequestrated lobes in the three cases were by large tortuous arteries arising from the abdominal aorta traversing the diaphragm via the inferior pulmonary ligament and entering the sequestration at the lower end. A dual arterial supply from the lower thoracic and abdominal aorta was observed in patient 3. The efferent venous drainage was by means of 3–4 anomalous veins draining directly into the pulmonary vein and two patients had additional venous drainage to the inferior vena cava. Histopathological examination confirmed the diagnosis of extralobar pulmonary sequestration with the basal area showing cysts lined by ciliated cuboidal to columnar epithelium and supported by attenuated smooth muscle. The epithelial lining of these cysts had interspersed polypoid projections containing fibroelastic connective tissue. The appearance was consistent with Stocker type 2 congenital cystic adenomatoid malformation within a sequestrated extralobar lung segment. The sequestrated segments were enveloped in their own visceral pleura (mesothelium-covered fibrous connective tissue similar to normal lung) and consisted of developed alveoli, bronchioles, and bronchi. The interstitium was intensely congested with the presence of pulmonary macrophages within the lumen. Thick elastic laminae were observed in the large vessels confirming systemic artery configuration. The vessels were increased in number near the pedicle of the extralobar pulmonary sequestration. The specimen of patient 3 who presented with pneumonia at 10 months of age showed bronchial dilatation, mucous plugging, and focal lipoid pneumonia. There was no evidence of inflammatory changes in the specimens of the other patients.
Discussion
The term “bronchopulmonary foregut malformations” was introduced to describe and explain intralobar/extralobar pulmonary sequestration associated with a communication with the gastrointestinal tract that had involuted, and also for complex anomalies that maintain a patent communication with the gastrointestinal tract due to incomplete involution.21 The spectrum of congenital bronchopulmonary foregut malformations includes intralobar pulmonary sequestration, extralobar pulmonary sequestration, congenital cystic adenomatoid malformation, bronchogenic cysts, and duplication cysts. These malformations probably share a common embryogenesis despite diverse morphology. In the fifth week of gestation the developing lung bud with its surrounding mesenchyme lies in close proximity to the primitive foregut from which it is derived. Differentiating lung epithelial cells, drawn by adhesions and chemotaxis towards the primitive foregut, may come to lie in the mesenchyme which is not conducive to their normal development, resulting in dysmorphogenesis of the misplaced tissue. Subsequent development of the pleura could sequestrate this ectopic tissue resulting in extralobar pulmonary sequestration. The accessory lung bud, if incorporated within the normal lung parenchyma, would lead to intralobar pulmonary sequestration. The tips of the primitive dividing bronchial buds are supplied by a systemic capillary plexus derived from the primitive aorta. If the plexus does not involute in response to the developing pulmonary artery an anomalous arterial supply may develop and exert traction on the ectopic tissue leading to sequestration. If the pulmonary artery continues to develop and supply the ectopic segment despite a disruption of the tracheobronchial connection, this may result in tracheopulmonary or bronchopulmonary malinosculation (pulmonary malinosculation simply describes a congenital abnormal connection or opening of one or more components of the bronchopulmonary vascular complex), producing such entities as congenital lung cysts and congenital cystic adenomatoid malformation. This hypothesis explains the related occurrence of pulmonary sequestration/congenital cystic adenomatoid malformation as seen in our cohort of patients.
Antenatal ultrasound screening permits early visualisation of fetal structural abnormalities and is a sensitive method of detecting pulmonary abnormalities. Sequential scanning has allowed the observation of the natural course of evolution of these anomalies. Despite the routine use of ultrasound there are no specific ultrasonographic features for reliable antenatal diagnosis of various lung anomalies such as congenital cystic adenomatoid malformation, pulmonary sequestration, or bronchogenic cysts. Antenatal diagnosis of pulmonary cystic abnormalities can be made as early as 18 weeks of gestation and sequential scanning has helped in deciding on fetal intervention, or in adopting conservative management. Serial antenatal scans have shown disappearance of 68% of lung lesions classified as sequestration which were later detected postnatally by radiography, CT scanning or MRI scanning and, similarly, fetal cystic adenomatoid malformations are know to regress or increase in size.6 ,22-24
Antenatally, cystic adenomatoid malformations can grow causing mediastinal compression and fetal hydrops; 55–85% of congenital cystic adenomatoid malformations detected antenatally can present with neonatal respiratory distress requiring surgical resection.6 ,22-24 Congenital cystic adenomatoid malformations may present later in life as recurrent chest infections and have been diagnosed incidentally in asymptomatic children.25 There is also the potential for congenital cystic adenomatoid malformation to undergo malignant change.20 ,26
Antenatally diagnosed pulmonary sequestration often spontaneously involutes to become nearly undetectable at birth.27 ,28 Of the 38 cases reported in the English literature 13 (34%) were found to have involuted and two (5%) disappeared completely.22-24 ,29 ,30 Sequestration antenatally is known to cause mediastinal deviation. Mediastinal deviation was associated with polyhydramnios in 60% of cases, and polyhydramnios was a major factor leading to poor prognosis and fetal morbidity.22 Persistent postnatal pulmonary sequestration may present with recurrent infection or cardiac failure later in childhood. The development of high output cardiac failure from hyperdynamic circulatory state due to significant massive systemic arterial blood supply and left to right shunting is described in infants with intralobar pulmonary sequestration, although this is not reported in association with extralobar pulmonary sequestration.7 ,22 ,31 In 75% of cases the anomalous artery arises from the aorta whereas venous return is provided by a pulmonary vein.31 ,32 The artery may also originate from the subclavian, intercostal, phrenic, internal thoracic, coeliac trunk, or left gastric arteries.7 ,22 ,31 ,32 The anomalous systemic arterial supply is observed to be multiple in 15% (60% in our cohort), but is most frequently by a solitary vessel arising either low in the thoracic aorta or from the abdominal aorta.33It is essential that an accurate identification of the vascular supply be made in all cases of pulmonary sequestration and pulmonary sequestration/cystic adenomatoid malformation.
The sequence of radiological investigations postnatally should be ultrasound, Doppler, followed by CT or MRI scanning. These should accurately map the complicated vasculature and show any other associated changes within the sequestration, resulting in the surgery being less hazardous. The above sequence of investigations for delineating the vascular supply should be followed in all cases of bronchopulmonary foregut malformations to avoid intraoperative surprises. The aberrant arteries are elastic, as evidenced by histological examination in all our patients, and often have atherosclerotic changes which make them extremely retractile and friable. Hence, at surgery the vessels and the inferior pulmonary ligament must be suture-ligated with care.
Including our five cases, 41 cases of pulmonary sequestration/cystic adenomatoid malformation with a systemic arterial supply have been reported. Nineteen of these (46%) were intralobar pulmonary sequestration with type 2 or type 3 congenital cystic adenomatoid malformation, and 18 (95%) arose in the lower lobe; 22 (54%) have been reported as extralobar pulmonary sequestration with histological type 2 congenital cystic adenomatoid malformation in 21 (95%) of cases.6-20 Ten of the 19 cases of intralobar pulmonary sequestration/cystic adenomatoid malformation (53%) were diagnosed antenatally. The natural history appears to be dependent on the size of the mass and secondary physiological derangement. One patient with a large mass presented with fetal hydrops necessitating fetal surgical excision and one neonate died due to associated prematurity at 26 weeks gestation. Three cases (16%) developed respiratory distress needing resection in the neonatal period, four (21%) developed infection in the mass during the first four months of age, and 10 (53%) remained asymptomatic.6-14
In comparison, 14 of the 22 cases of extralobar pulmonary sequestration/cystic adenomatoid malformation (64%) were diagnosed antenatally. The antenatal natural history is dependent upon the location of the lesion in extralobar pulmonary sequestration/cystic adenomatoid malformation. Intra-abdominal extralobar pulmonary sequestration/cystic adenomatoid malformation is not associated with fetal sonographic features of polyhydramnios or fetal hydrops, which are predictors of fetal morbidity. Intra-abdominal extralobar pulmonary sequestration/cystic adenomatoid malformation was asymptomatic without associated morbidity or mortality. Four of the seven antenatally diagnosed intrathoracic lesions had polyhydramnios. Two patients died and in the two surviving fetuses the polyhydramnios resolved, the extralobar pulmonary sequestration/cystic adenomatoid malformation decreased in size, and the mediastinum returned to the midline.6 ,15-19
Sixty six percent of the cases of persistent postnatal pulmonary sequestration without congenital cystic adenomatoid malformation may cause recurrent infections and/or high output cardiac failure. Congenital cystic adenomatoid malformation not associated with pulmonary sequestration may present as neonatal respiratory distress syndrome or result in recurrent chest infections in later childhood or adult life and have the potential to undergo malignant changes. Surgical excision is therefore recommended. The natural history of intralobar pulmonary sequestration/cystic adenomatoid malformation appears to be more favourable than that of congenital cystic adenomatoid malformation without an anomalous blood supply. Intra-abdominal extralobar pulmonary sequestration/cystic adenomatoid malformation seems to have a good prognosis. It is difficult to predict which intrathoracic extralobar pulmonary sequestration/cystic adenomatoid malformation will lead to hydrops and which will regress and have a variable prognosis.
The natural history of antenatally diagnosed lung masses is variable and, while counselling, the parents should be informed of the observed lung changes but no specific diagnosis should be given. The differential diagnosis must be considered with a careful search for other abnormalities and a multidisciplinary input during the prenatal course is essential. This would cede to smooth and structured postnatal investigations and management.
Postnatally a careful search for systemic anomalous blood supply should be performed by Doppler or MRI scanning, keeping in mind the association of pulmonary sequestration with cystic adenomatoid malformation in the differential diagnosis of antenatal fetal lung masses.
Pulmonary sequestration/congenital cystic adenomatoid malformation with significant systemic arterial blood supply is most unlikely to involute. Complications such as respiratory distress, infection, intrathoracic bleeding, haemoptysis, cardiac failure, and the potential risk of malignancy necessitate surgical excision of this congenital pulmonary lesion. Early surgery may reduce morbidity and parental anxiety.
Acknowledgments
Figure 2 is the original artwork of Madan Samuel.
References
Footnotes
Funding: none.
Conflict of interest: none.