Article Text

Statistics from Altmetric.com
Asbestos causes progressive pulmonary fibrosis (asbestosis), pleural disease (effusion and pleural plaques), and malignancies such as bronchogenic carcinoma and malignant mesothelioma.1-3 Asbestos is a generic term for a group of naturally occurring hydrated silicate fibres whose tensile strength and resilient structural and chemical properties are ideally suited for various construction and insulating purposes. The toxic effects of asbestos depend upon the cumulative dose and the time since the first exposure. Asbestos related diseases typically occur after a 15–40 year latency period following initial fibre exposure. The two classes of asbestos fibres, serpentine and amphibole fibres, can each cause pulmonary disease. Serpentine fibres, of which chrysotile is the principal commercial variety, are curly-stranded structures whereas amphiboles (crocidolite, amosite, tremolite and others) are straight, rod-like fibres. Chrysotile accounts for over 95% of world asbestos consumption.4 Asbestos induced pulmonary diseases remain a significant health concern. In the United States over 30 million tons of asbestos have been mined, processed and applied since the early 1900s.1 Moreover, non-occupational asbestos exposure may originate from existing buildings that contain enormous amounts of the fibres.5 Finally, it is estimated that the cumulative total number of asbestos associated deaths in the United States may exceed 200 000 by the year 2030.6
Extensive investigations over the last two decades have revealed some of the pathogenic mechanisms of asbestos pulmonary diseases. A further benefit of these studies is that asbestos induced pulmonary toxicity is an excellent paradigm to explore the mechanisms underlying other common causes of pulmonary fibrosis and malignancy. Asbestos is an established genotoxic agent that can induce DNA damage, gene transcription, and protein expression important in modulating cell proliferation, cell death, and inflammation.1 ,2 ,7-9 Recent comprehensive reviews have described in detail the histopathological and clinical features of asbestos related diseases1-3 ,9 ,10 as well as the evidence implicating various pathogenic pathways of asbestos induced lung diseases including (a) the chemical and structural properties of the fibres,1 ,2 ,7-9 (b) the lung fibre burden,9 (c) fibre uptake by pulmonary epithelial cells,11 (d) iron catalysed free radicals,8 ,12 (e) DNA damage,13 ,14 (f) cytokines/growth factors,7 ,9 ,15 and (g) cigarette smoke.8 ,9 Notably, no single mechanism fully accounts for all the complex biological abnormalities caused by asbestos. Moreover, the precise pathogenic pathways involved and their regulation are not fully established. In this review we focus on the important new information that has emerged over the last several years concerning the molecular mechanisms of asbestos related diseases. The primary goal of this review is to re-examine the evidence addressing the hypothesis that free radicals, especially iron-catalysed reactive oxygen species (ROS), have an important role in inducing pulmonary toxicity from asbestos exposure.
The amphibole hypothesis
The structural properties of asbestos fibres have been the focal point of theories of the pathogenesis of asbestos induced diseases.1-3 7-9 Amphibole fibres may be regarded as more toxic than chrysotile but this is an area of considerable disagreement.1 ,4 ,7 ,9 Compared with chrysotile, amphibole fibres accumulate more readily in the distal lung parenchyma, are not cleared as effectively, and are more durable (estimated half life in the lungs on the order of months versus decades, respectively).1 ,7 ,9 ,12 Chrysotile is also more readily dissolved after exposure to 4 M HCl for 30 minutes than crocidolite or amosite (60%, 6%, and 8% dissolution, respectively).12The structural features of amphiboles probably contribute to their greater biopersistence in lung tissue and hence their pathogenicity. Studies in humans reveal that amphiboles such as tremolite may contaminate chrysotile asbestos and contribute to the pathogenicity observed in occupationally exposed workers.1 ,9 Chrysotile induced asbestosis typically requires a threefold higher lung fibre concentration than amphiboles, yet parenchymal and pleural cells appear equally sensitive to chrysotile in terms of inducing asbestosis and mesothelioma in humans.9
Although fibre length is important in the fibrogenic and malignant capacity of asbestos in animal and in vitro models, human studies are less impressive.9 ,16 ,17 Davis and coworkers18 noted that rats that inhaled short amosite (>99% of the fibres <5 μm) for 12 months had negligible fibrosis whereas rats that inhaled long amosite (11% >10 μm) had extensive fibrosis. Hart and associates19 found that fibre length, but not diameter, directly correlated with fibre toxicity in Chinese hamster ovary cells in vitro such as inhibition of proliferation, induction of nuclear changes, and viability. As recently reviewed,11 there are conflicting data concerning the relationship between fibre size and uptake by pulmonary parenchymal cells. Furthermore, an association between fibre size parameters and the development of asbestosis in humans is unclear.9 ,11 ,16 ,17 The discrepancy between animal and human data may in part be due to the confounding effects of cigarette smoke which reduces lung fibre clearance.9 Recent reviews have also questioned the amphibole hypothesis in subjects with asbestos induced lung cancer.20 ,21 Thus, the structural characteristics of fibres (length, diameter, aspect ratio) alone appear insufficient to account for the pulmonary toxicity of asbestos, although certain physical characteristics may partly contribute to lung injury.
The free radical hypothesis
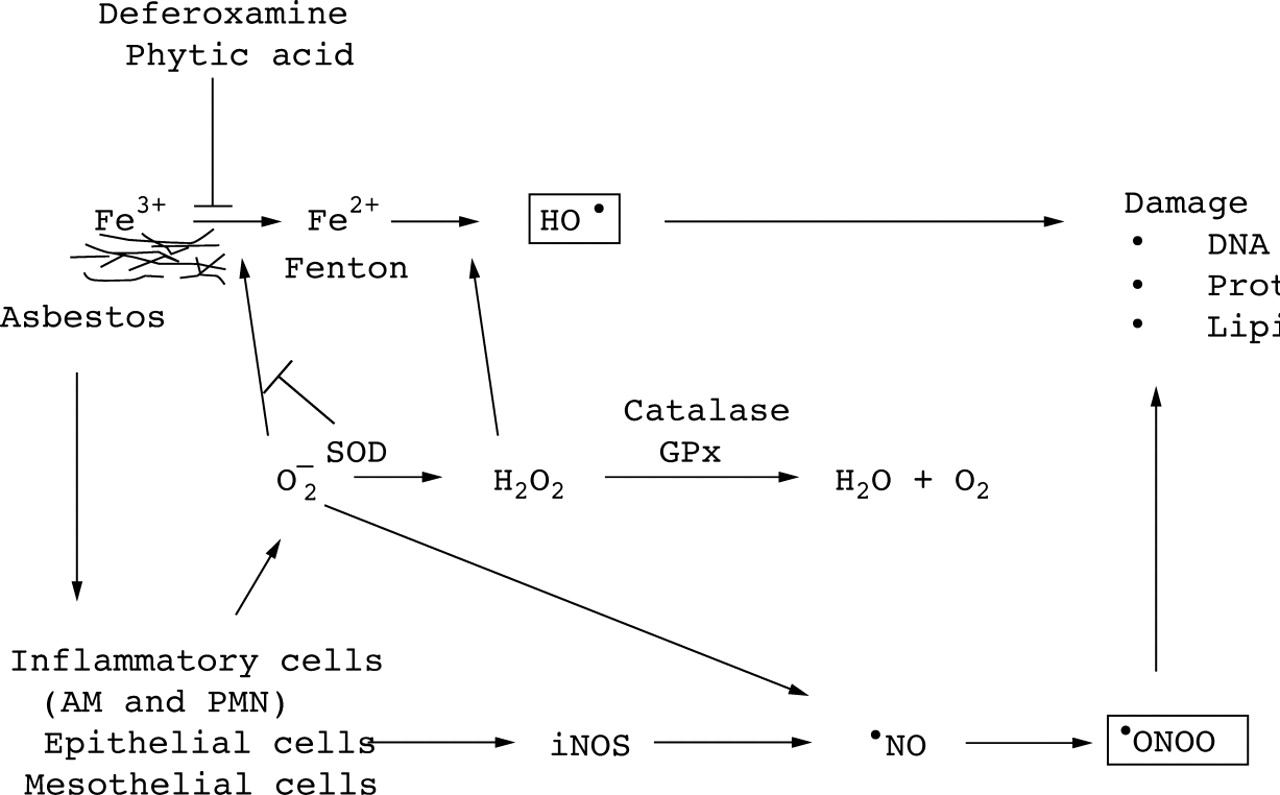
Considerable evidence suggests that ROS such as hydrogen peroxide (H2O2), superoxide anion (O2 –) and the hydroxyl radical (HO•), and reactive nitrogen species (RNS) are important mediators of asbestos toxicity.8 ,9 ,12 Asbestos produces ROS by at least two principal mechanisms. The first mechanism involves the iron content of the fibre augmenting HO• formation through iron catalysed reactions. The second mechanism implicates the release of ROS upon activation of inflammatory cells such as pulmonary alveolar macrophages and neutrophils. As reviewed below, recent studies have shown that asbestos also generates RNS such as nitric oxide (•NO) and peroxynitrite (•ONOO). Reactive oxygen species, especially HO•, and RNS, especially •ONOO, can alter biological macromolecules including proteins, cell membrane lipids, deoxyribonucleic acid (DNA), and ribonucleic acid (RNA) resulting in cellular dysfunction, cytotoxicity, and possibly malignant transformation.8 ,9 ,12 A hypothetical scheme delineating the free radical pathways is shown in fig 1.

Mechanisms of asbestos induced free radical production. The figure shows the hypothetical mechanisms by which asbestos stimulates the formation of reactive oxygen species and reactive nitrogen species as well as the relevant antioxidant defences. See the text for a detailed explanation of each pathway. Asbestos induced free radical production results from both direct (e.g. fibre) and indirect (e.g. inflammatory cell recruitment) mechanisms. AM = alveolar macrophage; PMN = neutrophils; H2O2 = hydrogen peroxide; O2 – = superoxide anion; HO• = hydroxyl radical; •NO = nitric oxide;•ONOO = peroxynitrite; iNOS = inducible nitric oxide synthase; DNA = deoxyribonucleic acid; Fe2+ = ferrous iron; Fe3+ = ferric iron; SOD = superoxide dismutase; GPx = glutathione peroxidase.
We review the evidence implicating ROS and RNS as mediators of asbestos pulmonary toxicity. Special emphasis is given to studies exploring the hypothesis that asbestos induced free radicals activate signalling cascades and cause DNA damage that results in altered gene expression and cellular toxicity important in the pathogenesis of asbestos associated pulmonary diseases.
GENERATION OF ROS IN CELL FREE SYSTEMS
The chemical structure of asbestos fibres can augment the formation of ROS in cell free systems. All types of asbestos have iron cations as an integral component of the crystalline structure, as a substitute cation, or as a surface impurity.12 ,22Amphibole fibres such as crocidolite (Na2[Fe3+]2[Fe2+]3Si8O22[OH]2) and amosite ([Fe,Mg]7Si8O22[OH]2) typically have a high iron content (∼27%) whereas chrysotile (Mg6Si4O10[OH]8) has a lower but significant iron content (∼1–6%), primarily as a surface contaminant.8 ,12
The iron associated with asbestos promotes the formation of the highly reactive HO• from H2O2.8 ,12 ,23 The Fenton reaction (equation 1), which is the primary equation involved in HO• formation, entails H2O2induced oxidation of ferrous iron (Fe2+) to ferric iron (Fe3+). Superoxide and other biological reducing agents can reduce Fe3+ iron back to redox active Fe2+ iron (equation 2). The chain of reactions in which H2O2 is converted to HO• is called the iron catalysed Haber-Weiss reaction (equation 3). Iron can also catalyse alkoxyl radical production from organic hydroperoxides as shown in equation 4. Ferrous iron can also induce the formation of other highly reactive free radicals such as ferryl (FeO2+) or perferryl species via a Fenton reaction.
Fe2+ + H2O2 → Fe3+ + HO– + HO• (1)
Fe3+ + O2 – → Fe2+ + O2 (2)
O2 – + H2O2 →iron→ HO– + HO• + O2 (3)
Fe2+ + ROOH → Fe3+ + RO• + HO– (4)
Electron spin resonance (ESR) spin trapping methods revealed that ROS are produced by asbestos in cell free systems. Weitzman and Graceffa used the spin trap 5,5′-dimethyl-1-pyrroline-N-oxide (DMPO) to show that chrysotile, amosite, and crocidolite asbestos each catalyse HO• production in the presence of H2O2.23 An important role for iron was suggested by the finding that an iron chelator, deferoxamine, inhibited HO• formation. These data have been corroborated by others and extended to include a wide variety of natural and man made mineral fibres.8 ,12
The location of the sites responsible for catalytic reactivity of asbestos is currently unknown. Aust and coworkers12 ,22found that the rate of mobilisation of iron from asbestos and its reactivity in cell free systems depends upon the pH and the chelator used (pH 5 > pH 7; EDTA > deferoxamine > citrate).12 ,22 Deferoxamine complexes with Fe3+ iron rendering it redox inactive, whereas EDTA mobilises iron that is potentially redox active resulting in HO• and DNA single-strand break (DNA-SB) formation.12 Utilising an electrochemical method, it has been shown that the total amount of redox active iron on the surface of crocidolite and amosite was 4.3 (0.7) and 3.3 (0.7) nmoles iron/mg, respectively.46 Moreover, they observed that the iron in asbestos can be repeatedly oxidised and reduced. In contrast, Gold and associates24 found, using atomic absorption spectroscopy and inductively coupled plasma atomic emission spectroscopy, that various chelators remove only approximately 1–5% of the total iron on the surface and that this did not affect the ratio of Fe3+to Fe2+ ions in crocidolite and amosite asbestos (70:30 and 60:40, respectively) or alter the content of redox active Fe2+ at the surface. These observations may account for the limited efficacy of iron chelators in some bioassays described below.
“Catalytic” or “free” iron consists of two components: redox active and diffusible. Compared with Fe2+ in biological systems at neutral pH, Fe3+ is more stable but has a very low water solubility.25 Low molecular weight chelators—for example, citrate, ADP, ATP and GTP—can promote “free” iron at physiological pH by maintaining at least one of six iron ligand binding sites available for Fenton reactivity.22 ,25 Notably, O2 – can release catalytically active iron from binding proteins such as lactoferrin, transferrin, ferritin, and hemosiderin as well as from asbestos.25 ,26 HO• induced DNA damage occurs either by hydrogen abstraction or addition of HO•to the DNA.27 Iron catalysed Fenton reactions induce site specific DNA damage that depends upon the location of iron in either (1) the extracellular medium, (2) closely associated with DNA bases, or (3) loosely associated with DNA.28 Scavengers of HO• may, not surprisingly, have limited protective effects in cells exposed to asbestos, partly due to the limited access of these scavengers to the site of HO• formation and because HO• is highly reactive with an extremely small diffusion distance (∼6.9 nm).27
Asbestos and other respirable fibres acquire iron on their surface to form asbestos bodies by mechanisms that are poorly understood. The iron coating is redox active and can induce DNA-SB formation.29Hardy and Aust30 showed that redox active Fe2+iron binds to both asbestos and deferoxamine treated asbestos, although the latter is 20–50% less effective in binding iron. Ghio and associates31 demonstrated that asbestos fibres acquire redox active iron from the medium and that this is lessened by deferoxamine. They also observed that iron treated fibres injected into the pleural cavities of rats and recovered four days later had increased levels of iron bound to the surface. However, deferoxamine did not alter the iron binding capacity of the fibres or the inflammatory response in vivo. Thus, redox active iron can be derived from the fibre, the cells, and the surrounding medium. Further, the protective effects of iron chelators against the production of HO• and DNA damage in biological systems may be limited unless the chelator is continuously present at the site of specific Fenton-like reactions.
Iron catalysed free radicals may also partly explain the well described interaction between asbestos and cigarette smoking that increases the rate of bronchogenic carcinoma and, perhaps, pulmonary fibrosis.8 ,9 ,32 Jackson and colleagues33showed HO• spin adducts of DMPO and phage DNA-SB in solutions containing crocidolite asbestos and aqueous whole cigarette smoke. These investigators proposed a role for iron since (1) ferrous sulfate could substitute for asbestos to yield a similar amount of HO• spin adducts of DMPO and DNA-SB and (2) DNA damage was inhibited by iron chelators (1,10-phenanthroline or desferrithiocin). As reviewed elsewhere,8 Pryor and coworkers also found a direct correlation between the number of DNA nicks in circular closed DNA and the level of iron catalysed HO• spin adducts produced by crocidolite asbestos and aqueous cigarette tar solutions. We recently showed that amosite asbestos and aqueous whole cigarette smoke extracts induce DNA-SB formation in cultured alveolar epithelial cells that was, at least in part, due to the production of iron catalysed free radicals.34 Although these data firmly implicate iron derived free radicals, certain of the toxic effects of asbestos and cigarette smoke can also be mediated by free radicals produced by an electron transfer reaction that is independent of either iron or oxygen—for example, polynuclear aromatic cation radicals.8
There remain several questions about the role of iron in asbestos generating free radicals. It is uncertain where the redox active site is located and whether iron chelators reduce the catalytic effects of asbestos by removing iron in the crystalline structure, impurities on the surface, or both. Other metal ions in asbestos may also prove important to the catalytic properties of asbestos. The significance of iron and other metal ions that are leached from asbestos in causing pulmonary toxicity in vivo is not established. Chelators that block all the coordination sites of iron, such as deferoxamine or phytic acid, are effective inhibitors while those that leave sites open, such as EDTA, may actually enhance HO•generation.8 ,12 The protective effects of chelators are diminished over time in biological systems suggesting a dynamic flux between iron and the chelator or degradation of the chelator.
GENERATION OF ROS BY ASBESTOS STIMULATED INFLAMMATORY AND PARENCHYMAL CELLS
A second mechanism by which asbestos can augment lung ROS levels is by activating inflammatory cells recruited to the site of asbestos deposition. As reviewed elsewhere,8 asbestos stimulates the release of O2 –and H2O2 from alveolar macrophages and neutrophils. Vallyathan and coworkers35 used the erythrocyte sedimentation rate (ESR) and the spin trap phenyl-N-tertbutylnitrone to show that various mineral dusts promote the release of oxygen free radicals from human neutrophils and rat alveolar macrophages. A role for iron catalysed ROS was suggested by the finding that the iron chelators diethylenetriaminepenta-acetic acid and deferoxamine inhibited 80% of ROS generated by asbestos.35 Phagocytic cells exposed to asbestos in vivo promote basal ROS release or prime cells for greater ROS generation after exposure to a second stimulus.36 ,37 As recently reviewed,11 mineral fibres can also augment an oxidative burst and lipid peroxidation in pulmonary epithelial cells and fibroblasts that are phagocytising the fibres. Moreover, exogenous ROS resulting from cigarette smoke or ozone can augment asbestos uptake into these cells.11
Considerable evidence suggests that alveolar macrophages and neutrophils have an important pathogenic role in asbestos induced pulmonary toxicity. Alveolar macrophages protect the lungs by limiting the lung fibre burden. Notably, abnormalities of pulmonary gas exchange in asbestos workers directly correlate with the percentage of neutrophils in the bronchoalveolar lavage fluid, but not with the number or percentage of alveolar macrophages.38 ,39 Using an in vitro model we found that neutrophil derived H2O2 augments asbestos induced pulmonary epithelial cell cytotoxicity, and that the toxic effects of asbestos activated neutrophils are similar to those caused by H2O2 alone.40 An important role for neutrophil derived H2O2 in asbestos toxicity is also supported by the fact that polyethylene glycol (PEG) conjugated catalase, but not PEG conjugated superoxide dismutase (SOD), diminishes BAL fluid levels of neutrophils, lung injury, and fibrosis in a rat inhalation model.41 Leanderson and Tagesson42 assessed HO• formation based upon 8-hydroxydeoxyguanosine (8hOHdG) levels in mixtures of neutrophils and fibres and found that asbestos (crocidolite, amosite and chrysotile fibres) produced significantly greater levels of 8−OHdG than man made fibres such as rockwool, glasswool, and ceramic fibres (3–21 pmol versus 0.7–5 pmol 8−OHdG/105polymorphs, respectively). In contrast to neutrophils we noted that alveolar macrophages decrease asbestos induced alveolar epithelial toxicity in part because the macrophages release less H2O2 and are better able to sequester the fibres from the epithelial cells.43 However, alveolar macrophages from patients with asbestosis release increased levels of ROS, cytokines, and growth factors that may have a harmful effect on the lung.9
Although the mechanisms by which asbestos stimulates ROS formation from inflammatory cells are not completely elucidated, several important features have been identified. Compared with non-fibrous mineral dusts, asbestos fibres are generally better able to stimulate ROS release from phagocytic cells.8 ,9 Goodglick and Kane noted that short (75% ⩽1.0 μm) or long (83% >1.1 μm) fibres of crocidolite asbestos induce similar murine peritoneal macrophage mitochondrial depolarisation and H2O2 release when corrected for surface area.36 These data suggest that “frustrated” phagocytosis by alveolar macrophages and neutrophils alone is not the only mechanism by which ROS are released since short fibres (75% ⩽1.0 μm ), which can be completely engulfed by phagocytes, caused similar H2O2 release to long fibres.36 Asbestos fibres may also directly activate ROS producing enzyme systems such as nicotinamide adenine dehydrogenase (NADPH) oxidase and/or phospholipase C pathways with secondary activation of NADPH oxidase. The finding that asbestos increases phosphoinositol turnover in guinea pig alveolar macrophages44 and hamster tracheobronchial epithelial cells45 provides evidence in favour of this mechanism. Ishizaki and coworkers46 showed that crocidolite induced O2 – release from human neutrophils is attenuated by staurosporin, an inhibitor of PKC and NADPH oxidase. Further studies are necessary to define more clearly the mechanisms by which fibres augment ROS generation by phagocytic cells.
GENERATION OF RNS BY ASBESTOS
Increasing evidence suggests that •NO has an important role in modulating asbestos induced pulmonary toxicity. Nitric oxide is a universal signalling molecule that is produced enzymatically from arginine by constitutive or inducible nitric oxide synthase (iNOS) present in cells, as shown in equation 5.47 ,48
l-Arginine → O2 / NADPH / NOS → l-citriline + •NO (5)
Inflammatory cytokines and oxidant stress can each augment iNOS expression and activity in pulmonary alveolar epithelial cells.47 ,48 Peroxynitrite, a highly reactive oxidant formed during the interaction between •NO and O2 •, can attack a wide array of biological targets.47 ,48 Although precise mechanisms are controversial, •ONOO is a potent oxidant that may form HO• like free radicals by iron independent mechanisms.47 Notably, •NO may also have antioxidant functions since it attenuates H2O2induced lipid peroxidation and pulmonary artery endothelial cell injury as well as tert-butyl-hydroperoxide induced formation of oxoferryl free radical species.48-50
Asbestos induces the expression and activity of iNOS in alveolar macrophages,51 A549 cells,52 and mesothelial cells.53 Chao and coworkers52 demonstrated that •NO was required for crocidolite induced 8-OHdG formation in A549 cells. Janssen and associates54 showed that •NO generating systems, similar to asbestos, activate early response genes, c-fos andc-jun, activator protein-1 (AP-1) DNA binding, and apoptosis in rat lung epithelial cells. Quinlan and colleagues55 noted that alveolar macrophages from rats inhaling either crocidolite or chrysotile asbestos release increased nitrite (the stable oxidation product of •NO) and nitrate (a surrogate marker for •ONOO formation) levels and that this effect was reduced by the NOS inhibitor, NG-monomethyl-l-arginine (NMMA). Using an in vitro luciferase model, these investigators found that crocidolite asbestos as well as its non-fibrous analogue, riebeckite, activated the iNOS promoter. Choe et al 53demonstrated that, in the presence of interleukin-1β (IL-1β), chrysotile and crocidolite each increased iNOS protein expression in rat pleural mesothelial cells in vitro as well as the production of nitrite and nitrate. Expression of iNOS was not due to iron alone since carbonyl iron had a negligible effect. These investigators also found that asbestos (crocidolite or chrysotile) inhalation for one and six weeks augmented the formation of RNS and nitrotyrosine (a marker for•ONOO production) in rat lungs and pleura as assessed by immunohistochemistry and an ELISA assay.56 These data showing that asbestos activates iNOS expression in a variety of lung cells suggest that •NO derived free radicals mediate pulmonary toxicity. However, the aforementioned antioxidant functions of •NO suggest that there is a complex balance between the protective and destructive effects of RNS in asbestos induced pulmonary toxicity that requires further investigation.
CELLULAR TARGETS OF ASBESTOS INDUCED ROS
Pulmonary parenchymal cells including alveolar macrophages, pulmonary epithelial cells, mesothelial cells, endothelial cells and fibroblasts are all susceptible to the toxic effects of asbestos.7-9 ,11 The evidence implicating iron catalysed HO• in vivo is largely circumstantial, primarily because the highly reactive nature of these species enables investigators to identify only the “footprints” of past activity.8 ,25It is important to recognise that tissue destruction can also occur by ROS independent mechanisms such as inflammatory cell derived proteases and cationic proteins,57 cigarette smoke,8 ,32 and RNS as noted above. In this regard, asbestos increases lung epithelial cell permeability by mechanisms that appear to be dependent upon activation of tyrosine kinase signalling but that are independent of iron catalysed ROS.58
At least eight lines of evidence show convincingly that asbestos induced cellular damage is caused, in part, by ROS generated either by the fibre itself or by activation of phagocytes. First, Schapira and associates59 documented the presence of HO•in rat lungs one week after exposure to a single intratracheal instillation of iron loaded chrysotile asbestos. These data corroborate substantial in vitro data showing that asbestos generates HO•.
Second, as reviewed elsewhere,11 fibre uptake by the pulmonary epithelium is increased by ROS. Churg and associates60 ,61 found that cigarette smoke augments asbestos induced lung disease in guinea pigs and asbestos uptake by rat tracheal epithelial cells. A role for ROS was implicated based upon the observation that catalase, SOD, and deferoxamine reduce asbestos phagocytosis by tracheal epithelial cells.60 ,61 These investigators recently found that cigarette smoke caused a twofold increase in asbestos adhesion to rat tracheal epithelial explants that was reduced by pretreating the fibres with deferoxamine.62These data suggest a role for iron catalysed ROS in augmenting asbestos adhesion and uptake by the epithelium exposed to cigarette smoke. However, non-oxidant pathways involving serum proteins, especially vitronectin, are also important and suggest that fibre uptake into cells occurs by complex mechanisms that require further study.11
Third, in vitro and in vivo studies show that antioxidants, especially catalase and, to a lesser extent, SOD attenuate asbestos induced cytotoxicity in macrophages,36 ,63 ,64 pulmonary epithelial cells,45 ,65 mesothelial cells,36and human umbilical vein endothelial cells.66 Further, iron chelators protect against asbestos induced cell damage and rat lung fibroblast collagen production suggesting a role for iron catalysed ROS.36 ,45 ,63-68 We demonstrated that an iron chelator, phytic acid, reduces pulmonary inflammation and fibrosis in rats two weeks after a single intratracheal instillation of amosite asbestos.69
Fourth, antioxidant enzyme expression and activity in the pulmonary epithelium is augmented by asbestos which suggests that adaptive mechanisms are activated. Mossman and coworkers65 noted that crocidolite and chrysotile asbestos, but not glass fibres, increased total endogenous SOD activities in hamster tracheal epithelial cells after exposure in vitro for several days. These investigators showed that ROS produced by a xanthine/xanthine oxidase system also increased tracheal epithelial cell gene expression of manganese (Mn)-SOD but did not alter copper-zinc (Cu/Zn)-SOD, catalase, or glutathione peroxidase mRNA levels.70 Using an in vivo model, these investigators showed that asbestos induced pulmonary inflammation and fibrosis were accompanied by discordant increases in total lung steady state mRNA levels for manganese SOD and glutathione peroxidase (but not catalase or copper/zinc-SOD), as well as increases in lung antioxidant enzyme activities for catalase, total SOD, and glutathione peroxidase.71 In contrast to crocidolite, rats exposed to aerosolised chrysotile (0.18 and 8.2 mg/m3 air) for 3–20 days had less prominent pulmonary inflammation and fibrosis.72 Notably, high dose chrysotile increased Mn-SOD mRNA levels in this animal model.
Fifth, in vitro and in vivo studies have shown that asbestos induced ROS injure alveolar epithelial cells.40 ,58 ,67 Alveolar type II cells have a key role in repairing the damaged alveolar epithelium and in determining the extent of lung damage.73Using quantitative ultrastructural immunocytochemistry, Holley and associates74 showed that inhalation of crocidolite asbestos or silica increased Mn-SOD protein in the mitochondria of alveolar type II cells. Although antioxidant enzyme gene expression and activity in rat lungs and alveolar type II cells are increased after asbestos inhalation, it is inadequate to attenuate lung injury and fibrosis.41 ,71 This suggests that either the antioxidant defences in the lung are overwhelmed and/or other yet undefined mechanisms are important. The protective effects provided by exogenous PEG-conjugated catalase support the former hypothesis.
Sixth, Mossman and colleagues found that tracheal epithelial cell lines transfected with a murine Mn-SOD cDNA resulted in several clones of cells that had a marked increase in Mn-SOD gene copies, mRNA levels, and SOD activity that rendered the cells less susceptible to the cytotoxic effects of crocidolite.75 We recently transfected WI-26 (type I-like) cells with a catalase expressing plasmid and found that, compared with vector transfected cells, catalase overexpressing cells were significantly protected against both H2O2 – and asbestos induced DNA damage assessed by an alkaline elution technique.76 These in vitro data with epithelial cells are supported by some in vivo data reviewed above.41
Seventh, asbestos induces the expression of ferritin, an iron storage protein, presumably to limit cellular exposure to redox active iron. Aust and coworkers77 ,78 showed that uptake of crocidolite asbestos by A549 cells (malignant cells with type II-like features) increases iron levels in the cells that directly correlates with the induction of ferritin synthesis and the pathogenic effects of the fibre. These investigators suggested that asbestos induced A549 cell injury was caused by mobilisation of redox active iron resulting in the induction of ferritin synthesis as an adaptive protective mechanism. Silica also induces ferritin protein expression in human alveolar macrophages by a post-transcriptionally regulated mechanism.79 A role for iron catalysed ROS in this model is supported by the observation that deferoxamine blocked silica induced ferritin expression. Interestingly, enhanced sensitivity to H2O2 induced DNA damage occurs in cells expressing increased amounts of the human transferrin receptor which results in enhanced iron uptake.80 These data suggest that mutations at the regulatory sequences of the transferrin receptor can alter iron homeostasis and thereby promote mutations and malignant transformation in the cells exposed to ROS and asbestos.
Finally, asbestos fibres, but not non-fibrogenic particulates, cause apoptosis in mesothelial cells, alveolar macrophages, and alveolar epithelial cells.54 ,81-84 Apoptosis, or programmed cell death, is an important mechanism by which cells with extensive DNA damage are eliminated without inciting an inflammatory response.85 As reviewed below, the mechanisms underlying asbestos induced apoptosis are not established. However, iron catalysed ROS are probably involved, based upon inhibition of mesothelial and alveolar epithelial cell apoptosis by iron chelators and antioxidant enzymes.82 ,84
MOLECULAR TARGETS OF ASBESTOS INDUCED ROS AND RNS
The molecular targets of asbestos and its second messengers, ROS and RNS, include critical biological macromolecules such as lipid membranes, DNA, and signal transduction proteins.8 ,9 ,12A hypothetical model is depicted in fig 2. Asbestos mediated lipid peroxidation is one mechanism by which asbestos modifies cell membrane structure and function. A role for iron catalysed ROS in causing asbestos induced lipid peroxidation is suggested by the fact that iron compounds alone or as a constituent of asbestos catalyse formation of these products, and by the observation that antioxidants and iron chelators reduce lipid peroxidation.8 ,12 Lipid peroxidation products are also present in the plasma of asbestos exposed workers as assessed by thiobarbituric acid reactive substances (TBARS).86 However, the level of TBARS did not correlate with chest radiographic abnormalities. A causal relationship between lipid peroxidation and in vitro cell toxicity was also questioned by a study which showed that vitamin E inhibits asbestos induced TBARS production but not injury to mouse peritoneal macrophages.87 Thus, asbestos associated iron can catalyse the formation of lipid peroxidation products.

{kind=link}
{kind=link}
Mechanisms of asbestos induced pulmonary toxicity. This is a schematic illustration of the hypothetical mechanisms involved in asbestos induced pulmonary damage. See the text for a detailed explanation of each pathway. ROS = reactive oxygen species; RNS = reactive nitrogen species; H2O2 = hydrogen peroxide; O2 – = superoxide anion; HO• = hydroxyl radical; •NO = nitric oxide;•ONOO = peroxynitrite; DNA = deoxyribonucleic acid; MAPK = mitogen activated protein kinase; PKC = protein kinase C; TK = tyrosine kinase; FAK = focal adhesion kinase; IκB = inhibitory protein kappa-B; NFκB = nuclear factor kappa-B; AP-1 = activated protein-1; Mn-SOD = manganese-SOD; Cu/Zn-SOD = copper/zinc-SOD; GPx = glutathione peroxidase; APEX/Ref-1 = AP-endonuclease/redox factor-1; TNFα = tumour necrosis factor α; IL-1 = interleukin 1; TGF-β = transforming growth factor β; PDGF = platelet derived growth factor; IGF-1 = insulin-like growth factor 1; IL-8 = interleukin 8; MIP-1α = macrophage inflammatory protein 1α; MIP-2α = macrophage inflammatory protein 2α; MMP-1 = matrix metalloproteinase 1; KGF = keratinocyte growth factor.
Asbestos may also cause cellular toxicity by damaging DNA. Genotoxicity, an important step in neoplastic transformation, is generally attributed to any agent that alters the genetic material. The genotoxic capacity of asbestos was initially questioned because of its inability to cause unscheduled DNA synthesis, mutations, or strand breaks in cultured cells.88-90 However, as comprehensively reviewed recently,13 there is convincing evidence that asbestos causes DNA damage in cell free systems as well as in virtually all the relevant target cells such as pulmonary epithelial and pleural mesothelial cells. Asbestos induced DNA damage manifests as altered DNA bases, DNA-SB formation, apoptosis, chromosomal aberrations, and sister chromatid exchanges.
Asbestos induced DNA base pair alterations are probably caused by HO• and •ONOO since these oxidants commonly react with DNA to produce hydroxylated bases47 ,52 ,91 or DNA-SB.92 As reviewed elsewhere,8 ,13asbestos and man made fibres promote 8-OHdG formation in DNA in cell free systems. Iron catalysed free radicals derived from peroxides or organic hydroperoxides can also augment asbestos induced DNA damage in cell free systems.13 ,93 Asbestos induced DNA damage by iron derived HO• can also occur in relevant target cells as evidenced by the protective effects of deferoxamine94and phytic acid.95 In contrast, Fung and coworkers96 recently showed that crocidolite asbestos (2.5–10 μg/cm2) induced 8-OHdG formation in DNA isolated from rat and human mesothelial cells, and that this occurred by iron independent mechanisms since deferoxamine was not protective. Whether or not reduced glutathione (GSH) protects against asbestos induced DNA damage is also unclear. Crocidolite induced 8-OHdG formation caused a 3–4 fold increase in mutagenicity inSalmonella typhimurium and these effects were prevented by GSH.97 In contrast, we found that liposomal GSH and N-acetyl-l-cysteine (NAC), a reducing agent and glutathione precursor, did not prevent asbestos induced DNA-SB formation in A549 cells as assessed by alkaline elution.98 The mechanistic explanation for the variability in the above studies is unknown but probably relates, in part, to the relatively poor penetration of some chelators and reducing agents into the cell to the site of redox active iron formation within the cell, as well as to the different cell types and DNA damage assays used.8 ,12 ,13
Cellular apoptosis caused by DNA damage and other stimuli is likely to be one important determinant of the lung’s response to ROS and asbestos. Apoptosis is the major pathway responsible for the resolution of alveolar type II cell hyperplasia in acute lung injury.73 ,99 The balance between apoptosis and cell proliferation is important since failure of apoptosis may trigger an inflammatory reaction or the development of a malignant clone of cells, whereas exuberant apoptosis may promote epithelial barrier dysfunction and lung injury. Hagimoto and coworkers100 demonstrated that bleomycin induces apoptosis in mouse lungs as assessed by DNA laddering and terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labelling (TUNEL). Apoptosis occurred primarily in the bronchiolar and alveolar epithelium and was associated with the expression of FAS and FAS-ligand mRNA. This same group utilised TUNEL to show DNA strand breaks and apoptosis in the bronchiolar and alveolar epithelium of patients with idiopathic pulmonary fibrosis.101 Although the precise molecular controls of apoptosis are not established, ROS are one of the critical stimuli that induce apoptosis—either directly or as a second messenger after exposure to various agents.102-105 Oxidant induced DNA-SB formation is one well recognised mechanism that triggers apoptosis.85 Furthermore, antioxidant compounds and iron chelators prevent apoptosis after exposure to H2O2, ultraviolet irradiation, and IL-3 deprivation.102 ,104 ,105
Several groups have recently shown that asbestos, unlike non-fibrogenic particulates, induces apoptosis in mesothelial cells, alveolar macrophages, and alveolar epithelial cells (AEC).54 ,81-84 Although the mechanisms by which asbestos causes apoptosis in these in vitro models are not established, accumulating evidence suggests an important role for free radicals. Broaddus and coworkers82 demonstrated that catalase and deferoxamine each significantly reduced mesothelial cell apoptosis, which suggests a role for iron catalysed ROS. We also found that asbestos induced AEC (A549 and rat alveolar type II cells) apoptosis in vitro is reduced by HO• scavengers and iron chelators, but not by iron loaded phytic acid.84 Narasimhan and associates106 noted that, compared with primary cultured mesothelial cells, three mesothelioma cell lines were highly resistant to apoptosis caused by either asbestos or H2O2. Moreover, these differences were not due to alterations in the expression of Bcl-2 and Bax, two proteins that modulate apoptosis. One explanation why mesothelioma cell lines are more resistant than non-transformed cells is that human mesothelioma cell lines have increased Mn-SOD and catalase mRNA levels and activity that render the cells more resistant to the cytotoxic effects of an oxidant stress.107 Notably, cells transfected with Mn-SOD are resistant to apoptosis caused by tumour necrosis factor α (TNF-α), H2O2, and irradiation.108 ,109These data suggest that asbestos induced ROS play a critical role in mediating apoptosis and that increased activity of antioxidant defences, especially Mn-SOD and catalase, account in part for the resistance of malignant mesothelioma cells to apoptosis. Since malignant mesothelioma is highly resistant to chemotherapy and radiation,2 future studies are needed to define better the molecular mechanisms modulating asbestos induced apoptosis that could form the basis of novel therapy.
Caspases (ICE family) are a group of cysteine proteases—for example, interleukin 1β converting enzyme (ICE), CPP-32/YAMA, and apopain—that have an important role in mediating apoptosis caused by a variety of agents including mineral dusts.85 ,110 Caspases cleave poly-ADP-ribose polymerase (PARP) as well as other molecules such as DNA protein kinase, lamin B1, and topoisomerase. PARP may be particularly important since the onset of ROS induced apoptosis is closely associated with the production of PARP cleavage products and reduced PARP activity may impair normal cellular DNA repair mechanisms.85 ,105 PARP, a 116 kDa nuclear enzyme, is activated by DNA strand breaks and subsequently ADP-ribosylates various proteins, including itself. Prolonged PARP activation can deplete cellular NAD and ATP levels and thereby augment cell death.111 PARP ADP-ribosylates DNA-polymerase α and topoisomerase I, suggesting that PARP also plays a critical role within the regulatory apparatus as a molecular nick sensor controlling proliferation.112 Niacin, which inhibits PARP and maintains cellular NAD levels, reduces pulmonary fibrosis caused by bleomycin113 and paraquat.114 Selective activation of PARP accounts for differences in the inflammatory and fibrotic response to bleomycin in two strains of mice.115PARP activation is also implicated in mediating asbestos induced mesothelial cell apoptosis and pulmonary epithelial cell injury since the PARP inhibitor 3-aminobenzamide (3-ABA) is protective.67 ,82 Furthermore, we noted that phytic acid and catalase each attenuated asbestos induced PARP activation in cultured AEC, suggesting a role for iron catalysed ROS.116However, a central role for ICE or PARP molecules in regulating apoptosis seems unlikely since knockout mice deficient in either ICE or PARP develop normally.117-119 The redundancy of caspases probably accounts for the fact that loss of any one member does not alter apoptosis.85 ,117-119
Asbestos catalysed HO• production and DNA base modification might be another mechanism whereby asbestos is both cytotoxic and mutagenic. McBride and coworkers,120 using the M13mp2 phage DNA mutation assay, found that Fe2+treated DNA, in contrast to untreated DNA, contained a 20–80 fold greater frequency of mutations and that these effects were ameliorated with catalase and SOD. They also found that mutagenesis arises principally from an increase in single base substitutions including (in descending order of frequency): G→C transversions, C→T transitions, and G→T transversions.120 These investigators postulated that Fe2+/oxygen induced DNA damage is non-random since a clustering of mutations at specific gene positions was noted. Carmichael et al,121 employing a highly sensitive 32P-postlabelling technique to detect DNA damage, showed that a Fenton-type system of copper (or iron) and H2O2 caused 10 specific lesions. Interestingly, their data suggest that these lesions result from intrastrand linking of specific adjacent DNA base pairs and are not due to base substitutions or adducts such as 8-OHdG. Hei and colleagues,122 using the AL human-hamster hybrid cell system, noted that chrysotile is a potent mutagen at the S1 locus of the human chromosome and that SOD and catalase decreased mutagenicity. Using hgprt–, gpt+ Chinese hamster V79 cells, Park and Aust123 showed that crocidolite (6 μg/cm2) caused a twofold increase in mutation frequency at the gpt locus which suggests, as others have,13 that asbestos induces multilocus deletions. They also noted a possible role for iron catalysed ROS and •NO since deferoxamine treated fibres induced no mutations at the gpt locus while crocidolite plus•NO generating systems increased the mutation frequency more than crocidolite or •NO generating systems alone or added together. This suggests that a mutagenic species is produced by the synergistic interaction between •NO and free radicals derived from the iron in asbestos. Thus, asbestos mutagenicity is probably due partly to ROS and RNS which cause multiple genotoxic effects including single DNA base substitutions, intrastrand linking, point mutations, and large chromosomal deletions.
Like ROS and DNA damaging agents, asbestos can modify cellular function by stimulating signal transduction cascades. For example, cells exposed to diverse stimuli such as ultraviolet light, irradiation, and H2O2 can each rapidly increase membrane bound tyrosine kinase (TK), protein kinase C (PKC), and mitogen activated protein kinase (MAPK) signalling cascades which subsequently can activate transcription factors such as AP-1 and nuclear transcription factor kappa-B (NFκB).124-128 MAPK family members such as extracellular signal regulated kinases (ERK1 and ERK2),c-jun-NH2-terminal protein kinases/stress activated protein kinases (JNK/SAPK) and p38 are activated by a variety of extracellular stimuli.127 A critical balance between activation of ERK and the JNK/p38 pathways may be particularly important determinants that can promote cell survival or apoptosis.129 Zanella and associates130showed that asbestos, but not its non-fibrous analogues, stimulated prolonged phosphorylation of ERK1 and 2 and increased ERK2 activity in rat pleural mesothelial cells. They also found that asbestos induced MAPK activation is attenuated by an inhibitor of the epidermal growth factor receptor (EGF-R) associated tyrosine kinase. These data show that asbestos can activate MAPK signalling pathways through the EGF-R in a manner similar to ionising radiation and H2O2.131 Jimenez and colleagues132 from the same laboratory demonstrated that asbestos induced rat pleural mesothelial cell apoptosis is due to activation of ERK, but not JNK/SAPK, while H2O2induced apoptosis was associated with activation of both signalling pathways. Notably, a role for iron catalysed ROS was suggested by the observation that catalase, NAC, or treatment of the fibres with an iron chelator (desferrioxamine) each reduced ERK activity. The molecular regulation of proliferative and death signals in cells is complex. For example, O2 – alone can mediate Ras induced cell cycle progression that is independent of MAPK and JNK/SAPK.133 Also, a reduction in TNFα induced O2 – caused by overexpression of Mn-SOD attenuates human breast cancer cell apoptosis by mechanisms that are directly correlated with a reduction in the activation of MAPK, JNK/SAPK and NFκB.108
NFκB is a tightly regulated transcription factor involved in the activation of a number of genes including cytokines, growth factors, adhesion molecules, and NOS as well as proto-oncogenes—for example,c-myc—involved in cell proliferation and apoptosis.134 Regulation of cytokine gene expression by NFκB is important since cytokines are not stored within cells. NFκB is a dimer consisting of two Rel family proteins (p50 and p65) that is activated in the cytoplasm by phosphorylation of its inhibitor proteins (IκBα, etc) resulting in IκB proteolytic degradation and subsequent dimer translocation to the nucleus where it binds to DNA. Three lines of evidence suggest that ROS regulate NFκB activation: (1) oxidants such as H2O2 activate NFκB, (2) agents activating NFκB such as TNFα, IL-1β, radiation, and endotoxin generate an oxidative stress, and (3) antioxidants (endogenous or exogenous) and iron chelators can decrease NFκB activation.134
Asbestos activates transcription factors in a variety of relevant pulmonary target cells. Janssen and coworkers,135 using transient transfection assays in hamster tracheal epithelial cells, showed that crocidolite asbestos causes prolonged dose dependent transcriptional activation of NFκB dependent genes. A role for ROS was suggested by their finding that NAC eliminated NFκB DNA binding. More recently these investigators exposed rats for five days to inhaled crocidolite or chrysotile and demonstrated a marked increase in p65 immunofluorescence in the bronchiolar and alveolar duct epithelial cells at the sites of initial fibre deposition.136 After 20 days of exposure, proximal airway epithelial p65 immunofluorescence returned to baseline levels while discrete cells in the alveolar ducts and interstitium (alveolar macrophages or type II pneumocytes) had enhanced reactivity. In contrast to epithelial cells, no clear activity was noted in mesothelial cells at any time point. Simeonova and colleagues137 ,138 showed that asbestos and H2O2 both activate NFκB and NF-IL-6 in A549 and normal human bronchial epithelial cells which, in turn, induce IL-8 and IL-6 gene expression and protein release. A role for ROS was suggested by their observation that HO• scavengers and NAC each attenuated asbestos or H2O2 induced NFκB and NF-IL-6 activity as well as IL-8 and IL-6 protein expression. Furthermore, asbestos induced IL-8 secretion—as well as NFκB and NF-IL-6 binding activity—were also decreased by inhibitors of PKC (staurosporin) and tyrosine kinase (genistein and herbimycin A) which suggests that phosphorylation by both signalling cascades is important in activating these transcription factors. These investigators also found a threefold increase in IL-6 levels in cells obtained from the bronchoalveolar lavage fluid in patients with asbestosis. Gilmour and associates139 showed that mineral dusts, especially long fibre amosite asbestos, increase NFκB and AP-1 transcriptional activity in rat alveolar macrophages by a mechanism that directly correlates with the free radical activity of the fibres (assessed by DNA damage) rather than fibre Fe2+ release (assessed using desferrioxamine), fibre Fe3+ release (assessed using ferrozine), or alveolar macrophage GSH depletion. Faux and Howden140 noted that asbestos induced NFκB and AP-1 transcription factor activation is reduced by vitamin E, an inhibitor of lipid peroxidation, as well as by 5,8,11,14 eicosatetraynoic acid, a lipoxygenase inhibitor, but not by indomethacin, a cyclo-oxygenase inhibitor. These data suggest that asbestos induced ROS, including HO• like species and/or lipid peroxides, activate NFκB, NF-IL-6, and AP-1 transcription factors in relevant target cells which, in turn, augment the inflammatory and fibrotic response to the fibres. Additional research is required to determine the precise free radicals involved in NFκB activation, their mechanism of action, as well as the factors regulating NFκB induced apoptotic and proliferative signals in various cell types in the lung.
Asbestos and asbestos derived free radicals can also act as a tumour promoter to augment cellular proliferation important in the development of a malignant clone of cells. ROS are known tumour promoters in part by inducing immediate early genes, c-fos andc-myc.141 Furthermore, like other tumour promoters, asbestos increases ornithine decarboxylase expression, the rate limiting enzyme in polyamine biosynthesis, and activates protein kinase C.142 Asbestos activates the AP-1 family of transcription factors, including homodimeric (Jun/Jun) and heterodimeric (Fos/Jun) factors derived from thec-fos and c-junproto-oncogene families.128 ,139 ,140 Asbestos and H2O2 both cause squamous metaplasia in hamster tracheal epithelial explants by AP-1 dependent mechanisms.143-145 Further investigations are necessary to determine the factors regulating the complex balance between ROS induced DNA damage that stimulates proliferative signals and mutagenesis as opposed to DNA damage that triggers cell death.
Tumour suppressor genes such as p53, Rb1, p16INK4a, p15INK4b, WT1, ATM, and NF2 guard the integrity of the cell’s genetic material by preventing clonal expansion, cell growth, and metastasis of cells with altered DNA to allow time for DNA repair or apoptosis after the onset of DNA damage.14 ,146 Although there is little information regarding the role of most of these tumour suppressor genes in the pathogenesis of asbestos related pulmonary diseases, increasing evidence suggests that alterations in p53 function are important. Mutations in the p53 gene, which is located in band p13 of chromosome 17, are present in over 50% of human cancers, especially lung cancers.14 ,146 As recently reviewed, accumulation of p53 protein in human lung carcinomas directly correlates with the asbestos content but the relationship between p53 and mesotheliomas remains unclear.14 ,147 ,148 p53 induces genes involved in apoptosis including Bax, KillerDR5, and others, as well as genes involved in cell cycle arrest (p21,WIF1, Cip-1), autoregulation of p53 (MDM2), and DNA nucleotide excision repair (GADD 45).14 ,146 Overexpression of p53, which generally signals abnormal p53 function, also occurs in pulmonary fibrotic disorders.14 ,100 ,101 Misra and associates149 recently demonstrated enhanced expression of p53 protein at the site of fibre deposition (bronchiolar-alveolar duct bifurcation) in rats exposed to aerosolised chrysotile. p53 immunoreactivity, which occurred primarily in alveolar macrophages and epithelial cells at the bronchoalveolar duct junction, peaked eight days after exposure and returned to basal levels by 30 days. Inactivated p53 is also overexpressed in cells infected with SV40 virus because p53 complexes with the large T antigen.14 As reviewed elsewhere,2 ,150 recent studies suggest a causal association between malignant mesothelioma and the presence of SV40 large T antigen in malignant mesothelioma cells. SV40 can also induce malignant mesotheliomas in experimental animals.150However, the significance of these provocative findings is uncertain and requires further studies involving a larger number of patients.
The levels of p53 mRNA generally correlate with the extent of DNA damage, while p53 protein levels can also increase by post-transcriptionally regulated mechanisms.146 Although the molecular mechanisms regulating p53 expression after DNA damage are not established, proteins recognising altered DNA as well as redox dependent mechanisms have been implicated.146 ,151 One putative p53 regulating protein is PARP since PARP deficient V79 fibroblasts are unable to induce p53 mRNA and are incapable of undergoing apoptosis.152 Since asbestos induces DNA damage and PARP activation in cultured mesothelial cells and AEC, p53 activation may be responsible for proliferation blockade and apoptosis that occurs in these relevant target cells.116 ,153 In this regard, chrysotile triggers mesothelial cell cycle arrest that is associated with increased p53 expression and an accumulation of cells in the G0/G1 phase of the cell cycle.153 These investigators also noted that chrysotile was more potent than crocidolite in these cells, and that each fibre induced a low level of apoptosis (⩽3%) suggesting that DNA repair mechanisms were activated. As reviewed by Broaddus,154 the choice of life or death after asbestos exposure probably depends on unique features of the exposed cell (for example, intrinsic antioxidant defences and DNA repair mechanisms), the extent of DNA damage, or external factors such as growth factor signals.
Redox conditions also modulate p53 activity but the role of asbestos in this regard has not been examined. Hainaut and Milner155showed that DNA binding of p53 is inhibited by a metal chelator and augmented by reducing agents. A role for a redox sensor regulating p53 expression in transformed cells was also suggested by the finding that sulphur containing antioxidants such as NAC, but not chain breaking antioxidants such as vitamin E, induced p53 mediated apoptosis.151 Thymidylate synthetase, which is a redox regulated protein, binds p53 mRNA and decreases p53 protein synthesis.156 Future investigations will undoubtedly provide better insight into these areas, in part by defining the genes involved in asbestos induced p53 expression as well as determining the significance of the interaction with SV40 virus exposure.
One important downstream target of p53 is the induction of p21. p21 expression can also occur by mechanisms that are independent of p53. p21 protein binds to a number of cyclins and cyclin dependent kinases, thereby inhibiting kinase activity and blocking cell cycle progression at the G1 checkpoint.157 Whereas p53 dependent mechanisms of p21 expression are closely associated with DNA damage, p53 independent mechanisms of p21 expression are triggered via a variety of pathways including cell senescence, TGFβ, and ROS.158-160 Hyperoxia induced proliferation blockade of alveolar type II cells appears to be mediated by a p53 dependent mechanism.161 ,162 A pathogenic role for p53 and p21 in pulmonary fibrosis is suggested by the finding of increased expression of both proteins in the bronchiolar and alveolar epithelium in patients with idiopathic pulmonary fibrosis.101 Chrysotile induced and crocidolite induced p21 expression in mesothelial cells causes a proliferation delay.153 Johnson and Jaramillo160 noted that crocidolite asbestos causes cell cycle arrest in A549 cells that correlates with the expression of p53, p21, and GADD153. One study showed that increased p21 expression occurs in 73% of human lung carcinomas and that this directly correlates with the five year survival independent of tumour staging or p53 expression.163 These data underscore the complexity of the interactions between p53 and p21 in the pathogenesis of pulmonary fibrosis and malignancies. Additional research is required to determine the role of asbestos induced free radicals in altering p21 expression and the subsequent impairment of cell proliferation.
Altered DNA repair mechanisms, which have recently been reviewed,13 ,164 may also be important in mediating asbestos pulmonary toxicity. The precise mechanism by which ROS and asbestos activate DNA repair pathways in eukaryotic cells is complex and not well established. It seems likely that cells exposed to asbestos will utilise repair mechanisms similar to those activated after exposure to ROS. H2O2 induced DNA damage is primarily caused by ROS derived from iron mediated Fenton reactions that occur at specific sites on DNA where iron is bound and, to a lesser extent, is also due to diffusible HO• or HO• like species generated by H2O2.164 Repair of H2O2 induced DNA damage mainly occurs by base excision but may also occur by at least three other pathways including direct restitution by hydrogen donation from a sulfhydryl, nucleotide excision, and recombination.164 DNA repair mechanisms are reduced in patients with lung cancer, especially in smokers.165 Notably, Lu and coworkers166showed that a human and murine 8-oxoguanine DNA glycosylase responsible for removing oxidatively damaged guanines resides, hOgg1, is located on chromosome 3 (3p25/26), a locus frequently deleted in cancers.
Abasic (AP) sites induced by oxidative free radical DNA damage are repaired in part by a unique AP-endonuclease—for example, APE and APEX—that contains a redox sensitive site (redox factor 1 (Ref-1)) located on its N-terminal portion.167 The human APEX/Ref-1 gene is located on chromosome 14q11.2-q12 and its promoter region has features consistent with a housekeeping gene.168 A 45 kDa protein that complexes with APEX/Ref-1 is crucial for the repair of H2O2 induced DNA damage.169APEX/Ref-1 is a potent activator of p53 by both redox dependent and redox independent mechanisms.170 Fung and coworkers171 showed that asbestos activates APEX mRNA, protein, and activity in primary isolated rat pleural mesothelial cells. Using dual fluorescent confocal microscopy, these investigators localised expression of APEX/Ref-1 to the nucleus as well as in the mitochondria. In contrast to asbestos, H2O2 did not induce APEX/Ref-1. The explanation for the distinct effects of various stimuli on APEX/Ref-1 expression is unclear but may be due to the cell type and experimental conditions. These data suggest that APEX/Ref-1 may be important in coordinating the redox state of the cell with DNA repair, p53 expression, and transcriptional activation.
Altered gene expression in cells that are chronically exposed to an oxidant stress probably contributes to pulmonary toxicity from asbestos. As mentioned above, antioxidant enzymes are increased in pulmonary epithelial cells and pleural mesothelial cells as well as in rat lungs exposed to asbestos. Gene amplification can increase antioxidant enzyme levels, especially catalase, in fibroblasts exposed to H2O2 (50–800 μM) or hyperoxia (80–95% Fio 2) over a six month period.172However, the relevance of gene amplification with asbestos exposure is unclear. The stress protein, heme oxygenase, is induced in human-hamster hybrid cells after an eight hour exposure to either crocidolite or chrysotile.173 A role for ROS was suggested by the finding that heme oxygenase expression was prevented by catalase or SOD. The glutathione-S-transferases, a class of conjugating enzymes involved in detoxification as well as the formation of sulphadipeptide leukotriene inflammatory mediators, may have a role in the pathogenesis of asbestosis.174 In a cross sectional study of 658 asbestos workers, Smith and coworkers174 found that a glutathione-S-transferase deficiency was a risk factor for the development of asbestosis. The investigators hypothesised that this increased risk was due either to a reduced ability to detoxify electrophiles or to altered leukotriene production.
ROLE OF CYTOKINES/GROWTH FACTORS
The role of cytokines, cytokine binding proteins, and growth factors in regulating disease expression in fibrotic lung disorders including asbestosis has been extensively reviewed recently.7 ,9 ,15 Some of the important cytokines and growth factors implicated in the pathogenesis of pulmonary fibrosis include IL-1, TNFα, transforming growth factor β (TGF-β), platelet derived growth factor (PDGF), and IL-8. These agents amplify cellular injury and activate fibroblast proliferation and collagen deposition. Although alveolar macrophages are considered the primary source of these proteins, increasing evidence suggests that pulmonary epithelial cells are also involved. For example, asbestos induces pulmonary epithelial cells to release IL-8 while TNFα exposure triggers epithelial cell release of macrophage inflammatory protein-1α (MIP-1α).137 ,175 A number of observations suggest that altered immune responses are also important in asbestos pulmonary toxicity.176 Here we focus on the evidence that free radicals modulate the expression of cytokines and growth factors. The paradigm emerging from these studies is that low level oxidative stress due to asbestos can activate signalling mechanisms and transcription factors which subsequently augment the synthesis of inflammatory and stress response proteins.
Accumulating evidence suggests that TNFα is a key cytokine involved in asbestos induced pulmonary toxicity.9 ,15 Alveolar macrophages from patients with asbestosis and idiopathic pulmonary fibrosis release increased levels of TNFα.177 Lung epithelial cells, especially hyperplastic type II cells in patients with idiopathic pulmonary fibrosis, contain high levels of TNFα.178 TNFα can increase alveolar type II cell MIP-1α mRNA levels which suggests that TNFα can promote pulmonary inflammation through its effects on epithelial cells.175Simeonova and associates179 demonstrated that iron catalysed free radicals are important in stimulating TNFα release from rat alveolar macrophages since deferoxamine or membrane permeable HO• scavengers each blocked TNFα release while ferrous sulphate or inhibition of intracellular catalase each augmented TNFα release. Using quantitative reverse transcription and polymerase chain reactions to assess mRNA levels for TNFα, they showed that alveolar macrophages exposed for 18 hours to asbestos had a 13-fold increase in TNFα mRNA that was completely blocked when asbestos was incubated in the presence of a HO• scavenger. The cellular effects of TNFα are mediated by two TNFα receptors (TNFR) that have a similar extracellular domain but vary in their intracellular domains and size (55 kDa vs 75 kDa). Using TNFR knockout mice, Brody and coworkers15 reported preliminary data showing that asbestos causes inflammation, cell proliferation, and fibrosis in the wild type and single TNFR knockout mice. Notably, asbestos caused no discernible damage in the TNFR knockout mice in which both TNFRs were not expressed. These innovative studies, if confirmed, provide compelling evidence that TNFα is a key proximal mediator of asbestos pulmonary toxicity. Furthermore, the available data suggest that successful management strategies could be developed by using antioxidants and/or iron chelators to reduce TNFα expression or by blocking TNFα binding to the TNFR. As reviewed elsewhere,180 there is some evidence to support the latter strategy in other model systems since administration of either anti-TNFα antibodies or soluble TNFα receptor inhibits fibrosis in mice exposed to bleomycin or silica.
Transforming growth factor β (TGF-β) represents a large superfamily of factors with diverse activities involved in growth and differentiation including pulmonary mesenchymal cell growth important in the pathogenesis of idiopathic pulmonary fibrosis and asbestosis.2 ,9 ,180 Many cell types synthesise TGF-β including alveolar macrophages, lymphocytes, fibroblasts, and pulmonary epithelial cells. Although there are multiple forms of TGF-β, most studies involving the lungs have focused on TGF-β1. The mechanisms underlying the fibrotic response of TGFβ are not established but probably involve (1) activating the expression of genes encoding for connective tissue matrix accumulation, (2) serving as a chemoattractant for fibroblasts and monocytes, (3) inducing its own transcription, (4) inhibiting the expression of genes encoding for proteases and antioxidant enzymes such as Mn-SOD, Cu/Zn-SOD and catalase, and (5) preventing the proliferation of epithelial cells.2 ,9 ,180 ,181 Khalil and coworkers182demonstrated that patients with advanced idiopathic pulmonary fibrosis have intense TGF-β expression in their bronchiolar and hyperplastic type II epithelial cells as well as in the subepithelial matrix. Sanatana and colleagues183 reported that bleomycin induces mRNA and protein expression of all three forms of TGF-β in alveolar macrophages and bronchial and alveolar epithelial cells. Corroyer and associates161 demonstrated that hyperoxia induced proliferation blockade of immortalised alveolar type II cells is due in part to increased type II cell expression of TGF-β1 which subsequently increases p21 levels. Whereas TGF-β can upregulate p53 activity by augmenting cyclin E levels,184 mutant p53 expression can abolish TGF-β induced growth inhibition.185 Perdue and Brody186 exposed rats to aerosolised chrysotile and showed intense staining for immunoreactive TGF-β1 in alveolar macrophages and low levels of staining in type II pneumocytes. Although the relevance of these in vitro findings with asbestos requires further study, the evidence suggests that asbestos induced free radicals induce TGF-β expression in relevant pulmonary cells. Also, several in vivo studies in other model systems have shown that systemic administration of antibodies to TGF-β attenuate extracellular matrix accumulation and fibrosis in several organs including the lungs.2
Keratinocyte growth factor (KGF), a fibroblast derived heparin binding mitogen, decreases oxidant induced pulmonary injury and mortality in animals exposed to hyperoxia, bleomycin, and radiation.187 ,188 Notably, KGF stimulates mesothelial cell proliferation in asbestos exposed animals which suggests an important role for this particular growth factor.189 The precise mechanisms by which KGF protects the lungs against oxidant injury are unclear. KGF may exert its protective effect by various pathways including (1) stimulating proliferation of alveolar type II cells to maintain barrier function,190 (2) reducing lung epithelial cell permeability,191 or (3) decreasing oxidant induced DNA damage by promoting DNA repair.192 ,193Further studies are necessary to determine the role of KGF in modulating lung injury caused by an oxidant stress such as asbestos, and to establish the mechanisms involved.
Conclusions
This review summarises some of the recent information concerning the molecular mechanisms underlying asbestos induced pulmonary disorders. Considerable in vitro and in vivo evidence suggests that free radicals, especially iron catalysed HO• and RNS, have a pivotal role in causing asbestos associated diseases. Confirmation of this hypothesis is difficult since free radicals, particularly HO•, are highly reactive and their destructive effects may have occurred years before disease presentation. The available data also suggest that the iron content of asbestos, as well as redox active iron associated with or mobilised from the surface of the fibres, is important in generating HO•. The evidence reviewed shows that asbestos induced free radical production is closely associated with the onset of DNA damage, signalling mechanisms, gene expression, mutagenicity, and apoptosis. The pathogenesis of asbestos induced diseases probably derives from the long term interplay between persistent free radical production and the expression of cytokines, growth factors, and other inflammatory cell products. However, the precise mechanisms by which asbestos and inflammation induced free radicals activate specific genes in pulmonary cells are not firmly established. Studies exploring the molecular basis of asbestos induced diseases are important for at least two reasons. Firstly, the development of effective diagnostic, preventive, and management strategies is predicated upon a firm understanding of the key pathways involved. Secondly, the asbestos model is a very useful paradigm for exploring the mechanisms underlying the production of free radicals, inflammation, fibrosis, and malignant transformation that are relevant to more common diseases such as lung cancer and pulmonary fibrosis.
Acknowledgments
This work was supported in part by a grant from the Veterans Administration (Merit Proposal). The authors appreciate the insightful comments from Dave Cugell.