Article Text

Statistics from Altmetric.com
Wegener’s granulomatosis (WG) is a necrotising granulomatous vasculitis which has a clinical predilection for the upper airways, lungs, and kidneys. The cause of WG remains unclear although recent investigations have begun to shed light on the immune mechanisms that may play a part in the pathophysiology of the disease. Therapeutic interventions in WG were largely ineffective until the introduction of combined treatment with cyclophosphamide and glucocorticoids which dramatically improved patient outcome. While disease relapse and drug toxicity remain of concern with this regimen, it has given us the opportunity for longer term follow up of patients from which we have gained a greater understanding of the disease.
Epidemiology
The prevalence of WG in the United States has been estimated to be at least three per 100 000 with a male to female ratio of about 1:1. Most patients (80–97%) are Caucasian, whereas African-Americans comprise about 2–8% of most cohorts. The mean age at diagnosis is about 40–55 years, although individuals of any age may be affected.1 Whether there is significant variation in onset of disease with different seasons or precipitation of illness by inhalation of certain particulate or gaseous materials or infection remains a matter of controversy. Recent studies have not supported the notion of seasonal variation whether the data be derived from patient perceptions of disease onset, medical records, or national hospital discharge surveys.1 ,2
A case-control study that compared patients with WG, healthy controls, individuals with idiopathic inflammatory pulmonary diseases and a variety of rheumatological diseases evaluated exposure to environmental particulate materials and gaseous substances.2 Over 75% of patients in all groups noted environmental exposure to inhaled materials during the year prior to onset of the diseases (or the interview date for healthy controls). The absence of a striking difference between patients with WG and control subjects, the rarity of familial cases, and the absence of a dominant HLA genotype among patients with WG support the notion that acquired host susceptibility plays an important part in the aetiology of the disease.
Clinical features
Over 90% of patients with WG first seek medical attention for upper and/or lower airways symptoms.3 Nasal and sinus disease is characterised by congestion and epistaxis due to mucosal friability, ulceration, and thickening. Perforation of the nasal septum and/or saddlenose deformity may occur from destruction of the nasal cartilage. Subglottic stenosis has been observed in approximately 20% of patients and can cause life threatening compromise of the airway. A high index of suspicion is important in detecting this lesion as it is frequently first diagnosed in the absence of other features of active WG and has non-specific presenting symptoms such as dyspnoea, voice changes, and cough.4 Although subglottic disease is optimally diagnosed by otolaryngological visualisation, an extrathoracic obstruction may be suggested by changes in the flow-volume loop (fig 1).

Flow-volume loop from a patient with Wegener’s granulomatosis with a subglottic diameter of 2 mm. Flattening is seen in both the inspiratory and expiratory phases consistent with a fixed extrathoracic obstruction.
In the lower airways WG can affect the pulmonary parenchyma, the bronchi, and rarely the pleura. Chest radiographs should be obtained in all patients suspected of having WG as as 34% of cases with radiographic abnormalities have been found to be asymptomatic.3 Although bilateral nodular infiltrates are common, a wide range of radiographic features can be observed including single nodules/infiltrates, cavitary disease, and diffuse alveolar haemorrhage (fig 2). Inflammation and stenosis of the endobronchial airways (fig 3) has been found to occur in at least 15% of patients with lung involvement.5 ,6 Endobronchial disease may present with cough, wheezing, dyspnoea, haemoptysis, or with symptoms related to lung collapse or post-obstructive infection.

Differing spectrum of radiographic abnormalities involving the pulmonary parenchyma in Wegener’s granulomatosis as seen by computed tomographic scanning: (A) bilateral nodular infiltrates, (B) cavitary disease, (C) pulmonary haemorrhage.
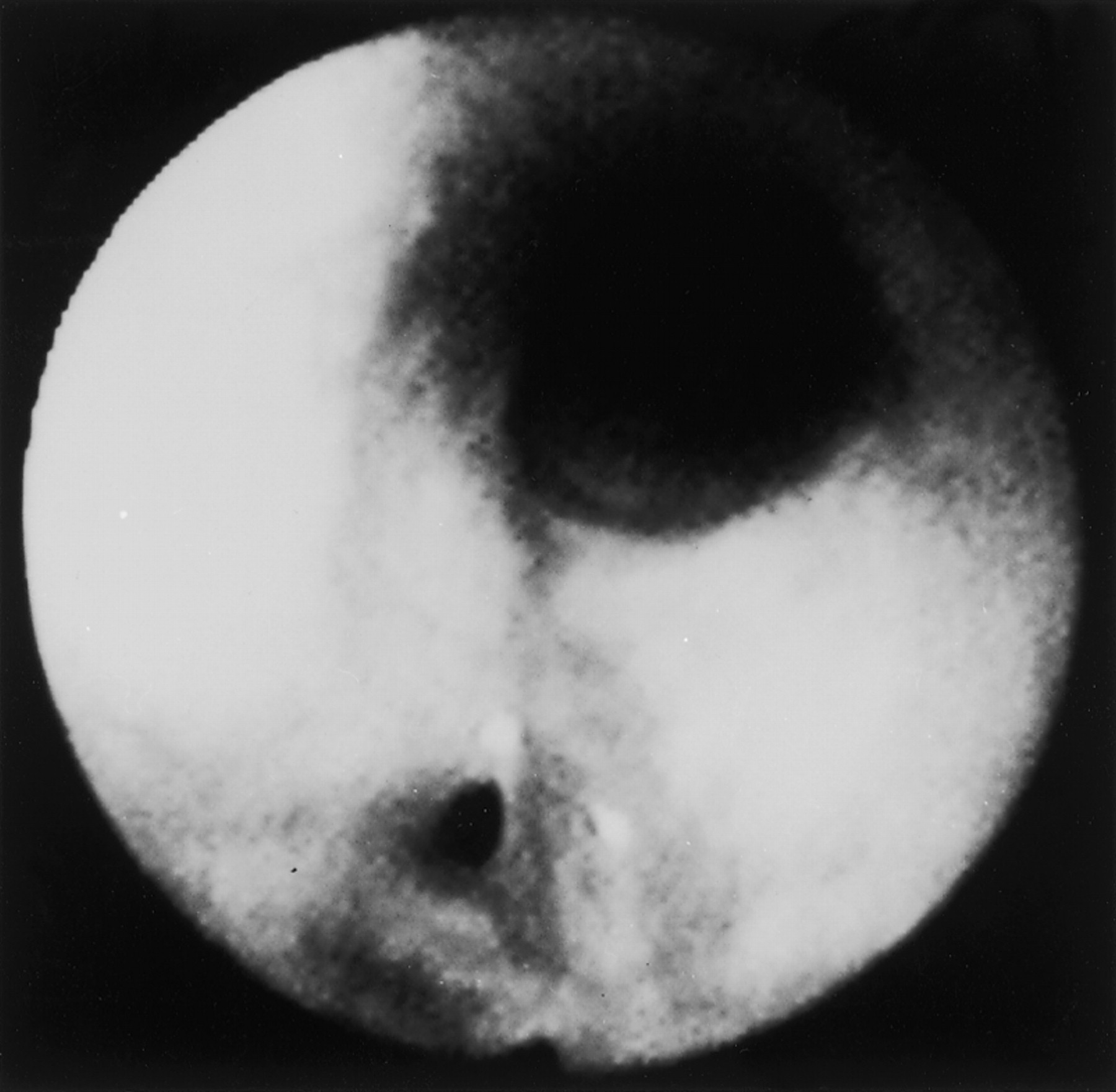
Narrowing of the endobronchial airways in Wegener’s granulomatosis.
Glomerulonephritis is one of the most serious disease manifestations of WG as it can progress rapidly to complete renal failure in the absence of symptoms.5 Detection is usually made by the presence of abnormal laboratory results such as proteinuria, an active urine sediment with microscopic haematuria and red cell casts, and potentially a decline in renal function as evidenced by a rise in serum creatinine or a decrease in creatinine clearance. Continued vigilance for glomerulonephritis is essential as it is present at diagnosis in only 20% of patients, but develops in 80% of patients at some point during the course of their disease.3
Although WG is classically thought of as involving the organ system triad of the upper airways, lungs, and kidneys, it can affect almost any site (table 1). Amongst the sites that have a substantial potential for morbidity are the eye and the nervous system. A wide spectrum of ocular manifestations has been observed in WG which may threaten vision by affecting the eye directly or by involvement of its contiguous structures.7 Such manifestations include episcleritis, scleritis, conjunctivitis, keratitis, uveitis, retinal vasculitis, optic neuritis, and retro-orbital pseudotumour. Mononeuritis multiplex has been observed in 15% of patients and 8% have had central nervous system disease.8
Wegener’s granulomatosis: clinical profile of organ involvement1-150
Diagnosis and clinical uses of ANCA
The differential diagnosis of WG is often based on the sites of organ involvement and frequently includes infections, neoplasms, connective tissue diseases, and other granulomatous diseases. As the medications used to treat WG are potentially toxic and could worsen these other conditions, a definitive diagnosis is essential.
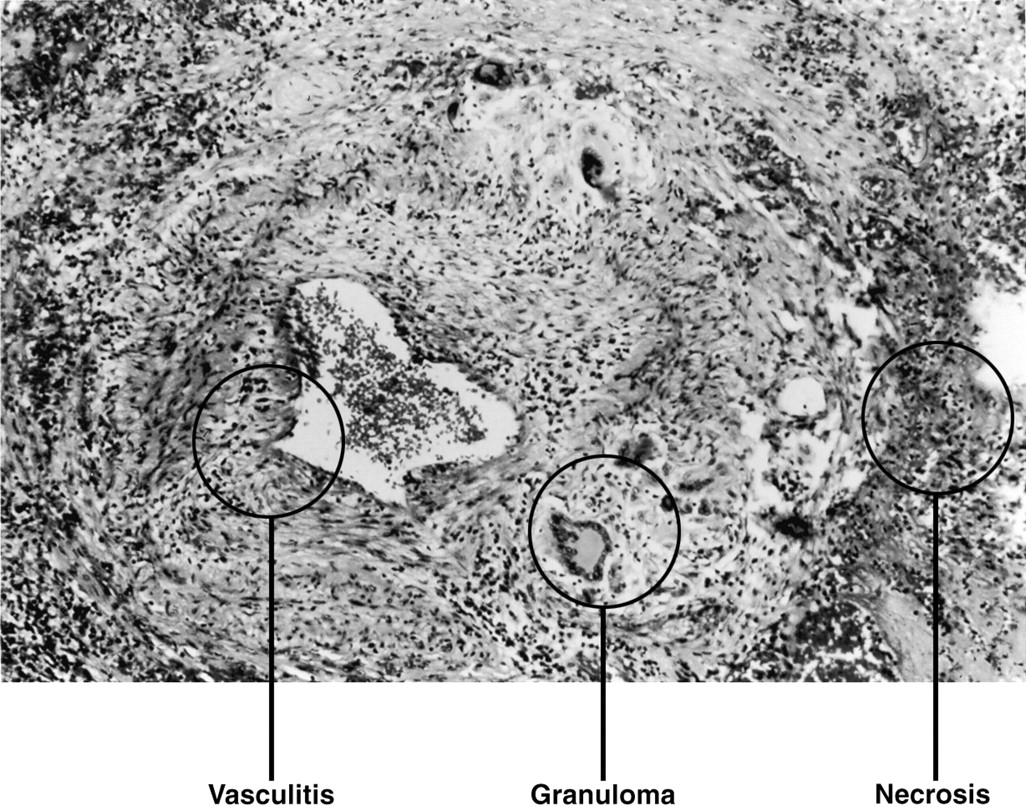
The diagnosis of WG is usually made by the histological demonstration of vasculitis, granulomatous inflammation, and necrosis9 in a clinically compatible setting (fig 4). These histological features are often patchy in distribution with the likelihood of obtaining a positive biopsy result being influenced by the organ site and, particularly, the amount of tissue obtained.10 ,11 The decision as to which site the biopsy sample should be taken from should be based on individual factors including the severity of illness, invasiveness of the procedure, likelihood of a positive yield based on data from the literature, and urgency of beginning treatment.

Pulmonary parenchyma showing the histological features of Wegener’s granulomatosis: necrosis, granulomatous inflammation, and vasculitis.
Antineutrophil cytoplasm antibodies (ANCA) were first described in 198212 and in 1985 were found to have a high degree of association with active WG.13 Since their description, two main immunofluorescent staining patterns of ANCA have been found—a cytoplasmic pattern (c-ANCA) and a perinuclear pattern (p-ANCA). c-ANCA has been found in 70–90% of patients with active WG; its target antigen is proteinase-3, a 29 kD serine protease found in the azurophilic granules of neutrophils.14 p-ANCA has a wider range of disease associations and antigen specificities but has been observed in 5–10% of patients with WG where it is usually directed against myeloperoxidase, another constituent of neutrophil granules.15 ,16
The sensitivity of ANCA for WG has been reported to range from 28% to 92%.13 ,17-19 This broad spectrum partly reflects the different criteria that were applied to establish the diagnosis of WG. Several series have demonstrated a high specificity of c-ANCA for WG ranging from 80% to 100%. This high degree of sensitivity and specificity of c-ANCA for WG has raised interest in whether this may be used as a means of diagnosis. Despite these findings, the usefulness of ANCA as a diagnostic test is also influenced by the pre-test probability of disease which will be low in most clinical situations given the uncommon nature of WG. Positive ANCA test results have also been reported to occur in other diseases that are part of the differential diagnosis of WG.16 ,20 Thus, while a positive c-ANCA test result may be useful in suggesting the possibility of WG, it should not normally be used in place of a biopsy sample to make the diagnosis.21
As ANCA titres will vary during the course of the disease and have been found to be higher in active than in inactive disease,13 ,17-19 some authors have questioned whether sequential ANCA titres can be used to predict disease relapse and guide treatment decisions.22 However, in one series in which patients were followed serially over 18 months, 44% of patients who had a fourfold rise in ANCA titre had no subsequent increase in clinical disease activity.23 Given the possibility that worsening disease may not occur and the toxicity of treatment may be considerable, a rising ANCA titre alone should not be used as the basis to reinstitute or increase treatment. Such a finding should, however, prompt a careful evaluation for any objective evidence of active disease and frequent patient monitoring.
Treatment
Untreated WG has had a poor outcome historically with a median survival time of five months.9 Glucocorticoids were the first treatment to be tried in WG and, while they delayed mortality, the median survival time remained at only 12.5 months.24Combined treatment with daily cyclophosphamide and glucocorticoids was an important advance.3 ,5 With this regimen patients received cyclophosphamide in a dose of 2 mg/kg/day for one year past remission which was then tapered and discontinued. Together with this, prednisone was initiated at a dose of 1 mg/kg/day. This dose was continued for one month after which time, if there was improvement, it was tapered on an alternate day schedule to discontinuation which usually occurred by 6–12 months. In 133 patients who received this treatment and who were followed for 6 months–24 years (a total of 1229 patient-years), 91% exhibited a marked improvement in their disease, 75% achieved complete remission, and an 80% survival rate was observed.3 This regimen remains the most effective treatment for active WG and any patient with immediately life threatening disease or rapidly progressive glomerulonephritis should be treated with daily cyclophosphamide and a glucocorticoid unless a strong contraindication exists. Despite the benefits of this treatment, disease relapse was found to occur in 50% of patients and morbidity from drug related toxicity was observed in 42%.3 In particular, transitional cell carcinoma of the bladder has developed in 6% of those treated with cyclophosphamide and has been projected by Kaplan-Meier estimates to have an incidence of 16% at 15 years following the patient’s first exposure to the drug.25Other malignancies seen with an increased frequency in cyclophosphamide treated patients include myeloproliferative disorders and a possible increase in skin cancers.3 ,26 Although some of the side effects of cyclophosphamide can be reduced by administering the drug intermittently, the prospective standardised trials that have been performed to date suggest that intermittent dosing is associated with a higher likelihood of relapse.27-29 For this reason it remains our recommendation that cyclophosphamide should be administered daily for WG.
The use of low dose methotrexate and glucocorticoids has been studied in patients who do not have immediately life threatening disease or who have developed cyclophosphamide associated toxicity.30-32 With this regimen patients received methotrexate 20–25 mg once a week plus prednisone which was tapered along a similar alternate day schedule to that described for cyclophosphamide. The methotrexate was given for one year after remission had occurred and was then tapered and discontinued. Of 42 patients treated with this regimen, 33 (79%) achieved remission with relapse occurring in 19 (58%) at a median of 29 months.32Only three patients failed to respond to the treatment and three fatalities occurred, none being due to active vasculitis.Pneumocystis carinii pneumonia was the most frequently observed severe side effect so we recommend prophylactic treatment with trimethoprim 160 mg/sulphamethoxazole 800 mg three times weekly for all patients with WG who are not allergic to sulphonamides and who are receiving a cytotoxic agent and daily glucocorticoids. These data support the use of methotrexate as an acceptable alternative for initial treatment in selected patients who do not have immediately life threatening disease or who have had significant cyclophosphamide related side effects. Methotrexate has also been found to maintain remission successfully in patients who were induced with cyclophosphamide.33 Although the follow up time in this series was too short to assess the relapse rate fully, it provides further evidence of the relative efficacy and safety of methotrexate in WG.
Data regarding other immunosuppressive and cytotoxic agents is limited, coming from case reports and small series. Such agents should only be considered as initial treatment where there are contraindications to cyclophosphamide or methotrexate. Azathioprine has not been found to be effective at inducing remission but may play a useful role in maintaining remission in patients who develop toxicity to cyclophosphamide.5 Chlorambucil and cyclosporin have had variable efficacy in reports from a small number of patients but their potential to cause malignancy and nephrotoxicity, respectively, needs to be considered.34 Although beneficial results with intravenous immunoglobulin have been reported,35 one series found no benefit in patients with major organ disease.36
Several authors have reported benefit from the use of trimethoprim/sulphamethoxazole in treating WG limited to the upper and/or lower airways,37 ,38 although methodological concerns have complicated the interpretation of these results. In a series of patients with biopsy proven WG in whom infection had been ruled out and other treatments had not been added or increased during the previous three months, trimethoprim/sulphamethoxazole was of little or no benefit in preventing disease progression.3Trimethoprim/sulphamethoxazole may be considered in patients with disease isolated to the upper airways but it is not recommended for treating lower airways disease and should never be used alone in the setting of glomerulonephritis. Whether trimethoprim/sulphamethoxazole can reduce disease relapse has also been investigated in a study by Stegeman et al 39 in which patients were randomised to receive the drug combination or placebo when their disease was in remission during or after treatment with glucocorticoids and cyclophosphamide. After 24 months 82% of the patients in the trimethoprim/sulphamethoxazole group were in remission compared with 60% of those who had received placebo. However, when broken down by type of relapse only the recurrence of upper airway disease was significantly reduced in the treated group with no difference being observed in major organ system relapses. As a cautionary note, although patients taking methotrexate can safely receive trimethoprim/sulphamethoxazole three times weekly for prophylaxis of Pneumocystis pneumonia, they should not be treated with trimethoprim/sulphamethoxazole twice daily as this combination has been associated with severe bone marrow suppression.32
While the presence of disease affecting a major organ (lung, kidney, nervous system) necessitates treatment with a glucocorticoid and a cytotoxic agent, not all manifestations of WG may require or respond to systemic immunosuppressive therapy. Isolated sinus symptoms may be treated with a stepwise strategy beginning with antibiotics, irrigation, and nasal steroids. If this is ineffective and infection is not thought to be present, the use of oral glucocorticoids and/or methotrexate may be considered. Patients managed in this manner must be monitored closely for evidence of disease activity in other organs as such involvement may be asymptomatic and would require a different therapeutic approach.
Subglottic stenosis related to WG frequently does not improve with medical treatment. One approach that has been found to be effective is to combine mechanical dilation of the trachea with intratracheal injection of a long acting glucocorticoid.4 Given the unresponsiveness of this lesion to medical treatment, patients who require immunosuppressive treatment for other manifestations should undergo this procedure concurrently. However, in the absence of major organ disease activity, WG related subglottic stenosis can be effectively managed using this technique alone.
Pathogenesis
Although the cause of WG remains unclear, several different lines of investigation have begun to probe the mechanisms which may play a part in the disease and provide possible areas for therapeutic intervention in the future.
In the past 15 years increasing circumstantial evidence supports the notion that ANCA may enhance the immuno-inflammatory events that contribute to WG and the related disorder, microscopic polyangiitis. Although the majority of patients with WG who make ANCA have antibody specificity to proteinase-3 (PR-3),40-42 not all patients are ANCA positive. In a study by Hauschild et al 40 more than 23 700 serum samples were tested from over 13 600 patients of whom 445 had WG. In those individuals who had limited WG without the presence of glomerulonephritis, ANCA was positive in 55%. In those with disease that included glomerulonephritis, however, ANCA was present in 88%. It appears that the more severe the illness, including the presence of glomerulonephritis, the more likely is the ANCA test to be positive. The important point from this study and many others is that ANCA is not universally present in all patients with WG. Attempts to identify immune complexes that contain antibodies to PR-3 and complement in affected tissues have generally been unrewarding. Thus, barring limitations in test sensitivity, the absence of ANCA in a significant minority of patients with active disease would indicate that, if it has a role, it is not an essential role. Nonetheless, a large amount of data indicates that ANCA, usually with specificity to PR-3 and occasionally to myeloperoxidase (MPO), may contribute to activation and injury of blood vessels.
Activated neutrophils express a variety of cytoplasmic antigens on their cell surface, many of which are enzymes.43-45 The binding of antibodies to displayed proteinase-3, MPO, and a variety of other neutrophil cytoplasmic enzymes enhances neutrophil activation, degranulation, and superoxide production. Neutrophil cytoplasmic enzymes may also bind to endothelial cells where they can produce injury directly, as well as enhancing endothelial cell activation.46 ,47 In in vitro studies the binding of anti-PR-3 to endothelial cells results in increased endothelial cell production of IL-8, an extremely potent neutrophil chemotactic agent.48 Concurrent with these events, endothelial cells exposed to anti-PR-3 also increase, in sequential fashion, IL-1α production and tissue factor, the principal initiator of the coagulation cascade.49 The monocyte, like the neutrophil, also produces PR-3 and, when activated and exposed to anti-PR-3, they will also increase IL-8 production markedly, further setting the stage for enhanced neutrophil chemotaxis.50 If endothelial cells were also capable of producing and expressing PR-3, further enhancement of vascular injury would follow. While some investigators have demonstrated PR-3 production by endothelial cells, others have not, and the conflicting findings are a matter of considerable controversy.51-54 The effects of ANCA that lead to enhancement of neutrophil and endothelial cell activation and injury are summarised in fig 5. In brief, these data represent powerful circumstantial evidence to suggest a role for ANCA in the pathogenesis of WG and microscopic polyangiitis. Although these observations demonstrate the potential for ANCA to enhance vascular injury, they do not explain organ selectivity. ANCA-based hypotheses would be more intriguing if studies that included preferentially affected organs in WG such as the airways and kidneys could be shown to provide a milieu that favoured ANCA mediated injury.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) When activated, neutrophils undergo degranulation and superoxide burst. Certain cytoplasmic antigens/enzymes can then bind to the neutrophil membrane including proteinase-3 (PR-3), bactericidal permeability increasing protein (BPI), myeloperoxidase (MPO), and lactoferrin (LF). When one of these antigens is bound by an appropriate antibody, such as anti-PR-3, the degree of neutrophil activation and superoxide burst is increased. (B) Following their release, proteins such as PR-3 may become bound to endothelial cells (ECs) where they may produce injury and endothelial cell activation. Antibodies bound to PR-3 on the endothelial surface may then lead to enhanced expression of adhesion molecules, increased production of IL-8 and neutrophil recruitment, binding and further endothelial cell injury. (C) Through these mechanisms a variety of pathways may be brought into play, all leading to vascular injury. At present, these mechanisms do not adequately explain preferential organ targeting in diseases such as WG where the greatest degree of injury occurs in the upper airways, lower airways, and kidneys. Reproduced from Hoffman and Specks42 with permission.
The potential role of infection in initiating and promoting the development of WG remains an area of intense interest. Patients with WG and other vasculitides often present with clinical features that suggest an infectious disease. In WG it is of particular note that the airway is almost always initially involved. Bronchoalveolar lavage studies have included patients with newly diagnosed disease and those who have experienced reactivation of disease, as well as individuals in remission. Even in those instances where clinical features of active airway disease are not present, patients generally have neutrophilic alveolitis at disease onset and disease reactivation.55-57 It has been suggested that airway stimulation, producing a neutrophilic response in an immunologically susceptible host, may trigger systemic events that we recognise as WG. To date, histopathological studies of airway biopsy specimens—including special stains for microorganisms and cultures for bacteria, mycobacteria, fungi, mycoplasma and respiratory viruses—have failed to reveal a causative infectious agent. However, most attempts have used conventional laboratory techniques. More sophisticated molecular techniques including polymerase chain reaction (PCR) have identified infectious agents in other diseases where culture requirements have been unusually fastidious and special tissue stains have been inadequately sensitive.
Further pursuit of an infectious aetiology should be encouraged for several reasons. Several infections are known to be associated with certain types of vasculitis. Marek’s disease, an accelerated form of atherosclerosis and vascular inflammation, is due to a chicken avian herpes virus. The illness can devastate entire flocks of animals and can be prevented by immunisation.58 In man, vasculitis has been reported to occur in association with hepatitis B, hepatitis C, Epstein-Barr virus, parvo-B19, and HIV infections. However, most patients who suffer from these infections develop a variety of clinical problems with only <1% of infected individuals developing vasculitis. This observation would suggest a unique host abnormality that leads to this special form of disease expression. Several animal studies have supported this hypothesis. For example, a variety of immunologically defective mice with the outward appearance of normality will develop vasculitis following gamma herpes virus infections. Vasculitis in these animals is particularly apparent in the setting of genetic deficiencies for gamma interferon or the gamma interferon receptor.59 ,60
Some have suggested that a persistent infection as a stimulus for vasculitis is not tenable because, in most instances, immunosuppressive therapies are life saving and patients do not die of overwhelming infection. Patients with hepatitis B or hepatitis C associated vasculitis often improve substantially, however, with immunosuppressive therapy while their viral load increases. A similar scenario is well documented in mink infected with the Aleutian disease virus. Different strains of viruses produce disease of differing severity and, although all mink are susceptible, Aleutian mink are particularly susceptible to infection. Infected animals produce circulating immune complexes and develop a lethal form of vasculitis which can be prevented by treatment with cyclophosphamide, although viraemia persists.61 ,62
These animal models in which vasculitis and infection occur together show similarities to vasculitis in humans. Organ and vessel involvement is usually non-uniform with numerous areas of “skip lesions”. Not all organisms produce the same pattern of vasculitis. In the mouse model with gamma herpes virus infection, disease occurs primarily in the large arteries such as the aorta and its main branches, whereas in Aleutian mink disease lesions are found primarily in small and medium sized vessels. It is possible that unique properties of infectious agents—such as affinities for specific tissue substrates, focal tissue properties, haemodynamic characteristics of an organ, and the unique immune response of different sites—may be important in vasculitis organ “targeting”. Recent studies in man suggest that infection with Chlamydia pneumoniae or cytomegalovirus may enhance the inflammatory component of atherosclerosis. Might these organisms also lead to an enhanced inflammatory response in an immunologically flawed host, similar to the scenario recognised in animals? It is our hope that at least some of the idiopathic vasculitic conditions will be found to have an infectious aetiology and thus the potential for curative therapy.
Another area of investigation has focused on trying to understand the mechanisms underlying the development of granulomas in WG. In other diseases granulomatous inflammation has been found to be a process mediated by sensitised CD4+ T cells that produce Th1 cytokines (IL-2, IFN-γ, TNF-α). The presence of similar inflammation in WG raises the question whether tissue injury and vasculitis may be mediated by an aberrant Th1 immune response. Support for this mechanism has come from several lines of research. Both quantitative and qualitative abnormalities in cytokine production have been described in WG and related vasculitic syndromes. Raised serum levels of IL-1, IL-2, IL-6, and TNF-α63 ,64 and increased production of TNF-α by circulating mononuclear cells65 have been reported in patients with WG. More recent studies have focused on the production of pro-inflammatory cytokines in active vasculitic lesions in situ. Increased production of IL-1 and TNF-α has been found in the renal glomeruli of patients with WG and active glomerulonephritis using reverse transcription polymerase chain reaction (RT-PCR), in situ hybridisation, and immunohistochemical techniques.66 Using a similar approach, Weyand and colleagues67 found mRNA for IL-1, TNF-α, IL-6, IL-2, IFN-γ, and TGF-β in sections of temporal arteries from patients with giant cell arteritis, another granulomatous vasculitis syndrome. In parallel studies of histologically normal temporal arteries from patients with polymyalgia rheumatica, mRNA for IL-1, TNF-α, IL-6, and IL-2 was detected but tissue samples did not contain IFN-γ sequences, suggesting that IFN-γ may be involved in the progression to overt arteritis.
Recent data by Ludviksson et al have provided the most compelling evidence to date that the T cells associated with the granulomatous inflammation of WG are skewed towards a Th1 cytokine pattern.68 Studies of peripheral blood lymphocytes in patients with active WG showed that CD4+ T cells produced 10–20 times higher levels of IFN-γ and significantly more TNF-α than did CD4+ T cells from normal controls. In contrast, there was no difference in the level of Th2 associated cytokines (IL-4, IL-5, or IL-10) produced by T cells from patients with WG compared with control subjects. The finding that WG lesions are associated with T cells skewed towards Th1 cell differentiation implies an abnormality in the regulation of IL-12, the primary inducer of T cells producing IFN-γ. Although the highest levels of IL-12 production were obtained with monocytes from patients with active disease, Ludvikssonet al observed that monocytes from patients with inactive disease also produced increased amounts of IL-12.68 This suggests that the increase in IL-12 production is not a secondary effect to the inflammatory process but, rather, a primary feature of WG.
Based on these observations, it has been hypothesised that exposure of patients with WG to environmental insults (such as infections) and/or autoantigens induces an excessive macrophage IL-12 response leading to an unbalanced production of Th1 cytokines. Such aberrant production of TNF-α and INF-γ could initiate and perpetuate the granulomatous inflammatory vascular lesion that characterises WG. This process may be further influenced by ANCA, which may enhance neutrophil, endothelial cell, and monocyte activation. However, as previously noted, because a significant minority of patients with WG are ANCA negative, an essential role for ANCA in this process is not likely. These findings have potentially important implications for the treatment of WG. In particular, they suggest that approaches which downregulate the Th1 pathway and IL-12 production may halt the inflammation. This is supported by the finding that the addition of exogenous IL-10 causes a dose dependent blockade of INF-γ production by peripheral blood mononuclear cells from patients with active WG.68
Conclusions
WG is a systemic vasculitis that can be associated with substantial morbidity and mortality as a result of the disease or its treatment. Much remains unknown about the factors which initiate and perpetuate the inflammatory process of WG, although significant advances have been made in recent years. As further understanding is gained, it might become possible to target therapeutically critical elements in the pathophysiological mechanisms underlying WG, thereby eliminating the risks of agents that are associated with non-selective immunosuppression. Until that time, however, treatment of active major organ threatening disease with a cytotoxic agent combined with glucocorticoids will remain the primary means of controlling the disease and prolonging patient survival.