Article Text

Abstract
BACKGROUND The importance of tumour necrosis factor-alpha (TNF-α) in the pathogenesis of pulmonary sarcoidosis has remained uncertain because of the paucity of clinical features associated with excessive levels of this cytokine. Increased levels of soluble TNF receptors (TNF-R), which are known to inhibit TNF-α activity, were recently described in the lungs of subjects with sarcoidosis. We hypothesised that TNF-α bioactivity may be inhibited in sarcoidosis by the presence of TNF-R. A study was therefore undertaken to investigate for the first time the relationship between soluble receptors and TNF-α bioactivity in the lungs of subjects with sarcoidosis.
METHODS Alveolar macrophages (AMs) from 16 subjects with histologically proven sarcoidosis and 13 healthy controls were cultured in the presence and absence of lipopolysaccharide (LPS). The subjects with sarcoidosis were grouped by radiological assessment into stage I (n = 6) and stage II/III (n = 10). The cell culture supernatants and bronchoalveolar lavage (BAL) fluid were assayed for TNF bioactivity using the WEHI 164 clone 13 assay. Immunoreactive (bound and free) TNF-α and free TNF-Rs (p55 and p75) were determined by ELISA.
RESULTS Bioactive TNF-α was undetectable in the BAL fluid of all the subjects with sarcoidosis and most of the healthy controls. However, there was significantly more immunoreactive TNF-α in the BAL fluid from subjects with sarcoidosis than from the controls (median values 0.304 ng/ml and 0.004 ng/ml, respectively, 95% CI 0.076 to 0.455, p<0.001). The levels of both p55 and p75 in the BAL fluid were higher in both sarcoidosis groups than in the controls (p<0.0005 and p<0.001, respectively). In LPS stimulated AM supernatants reduced TNF-α bioactivity was seen in subjects with stage I sarcoidosis compared with those with stage II/III disease and healthy controls (median 0.333 ng/ml vs 1.362 ng/ml and 2.385 ng/ml, respectively, p<0.01). This contrasted with increased p55 levels in the AM supernatants derived from subjects with stage I sarcoidosis compared with those with stage II/III disease and healthy controls (median 0.449 ng/ml vs 0.058 ng/ml and 0.078 ng/ml, respectively, p<0.01). The levels of p75 were increased in unstimulated AM cultures in subjects with stage II/III disease compared with those with stage I disease and healthy controls (median 0.326 ng/ml vs 0.064 ng/ml and 0.102 ng/ml, p<0.05).
CONCLUSIONS These results indicate that TNF-α bioactivity may be inhibited by increased soluble TNF-R in the lungs of subjects with sarcoidosis, and this inhibition may be greater in patients with stage I sarcoidosis than in those with stage II/III disease. This may represent a homeostatic mechanism which protects the lung from excessive TNF production characteristic of chronic inflammation.
- tumour necrosis factor
- sarcoidosis
- tumour necrosis factor soluble receptors
Statistics from Altmetric.com
Sarcoidosis is a multi-system disorder with 90% of patients exhibiting some degree of pulmonary involvement.1Pulmonary sarcoidosis is characterised by an accumulation of T lymphocytes in the alveolar spaces, preceding the development of non-caseating epithelioid granulomas which can lead to interstitial fibrosis in some cases.2 The clinical syndrome can be staged according to the appearance on the chest radiograph. Stage I disease is defined by the presence of bilateral hilar lymphadenopathy (BHL) with no interstitial changes, whereas stage II disease denotes BHL in association with nodular shadowing. Stage III is a more generalised shadowing in the absence of BHL, and stage IV disease classification requires radiographic evidence of fibrosis. Stage I sarcoidosis is usually a self-limiting disease, whereas stages II–IV may be associated with a more progressive clinical course.
Tumour necrosis factor alpha (TNF-α) is a pleiotropic pro-inflammatory cytokine which has been implicated in the pathogenesis of a number of inflammatory lung diseases3 ,4 by virtue of its ability to increase the respiratory burst in neutrophils5 and induce pulmonary vascular permeability,6 thus promoting inflammatory cell accumulation. Sarcoidosis has been described as an example of a disordered inflammatory response with increased production of a number of pro-inflammatory cytokines,7-11 but the role of ΤΝF-α in the pathogenesis of sarcoidosis remains uncertain. The documented production of γ-interferon and cholecalciferol in patients with sarcoidosis, both of which are extremely effective priming agents for TNF-α, suggests the potential for an active role if it is produced in this condition.12 Several studies have reported increased levels of TNF-α in patients with sarcoidosis,13-15 although this has not been a consistent finding.16 Indeed, many of the clinical and pathological features associated with TNF-α such as systemic disturbance, weight loss, and tissue necrosis are not commonly found in Caucasian subjects. This led to the hypothesis that TNF-α activity might be inhibited in sarcoidosis. In 1990 an inhibitory factor was described, but not characterised, in the circulation but not in the lungs of patients with sarcoidosis.17 Since then two forms of soluble TNF receptors (TNF-R) have been characterised from human urine18—the p55 (CD120a) and the p75 (CD120b) receptors. Both are identical to the membrane bound form and are cleaved enzymatically from the cell surface by a metalloproteinase enzyme.19 The p55 receptor is the main signalling receptor and is ubiquitous in human tissue, whereas p75 receptor expression appears to be restricted to cells of the myeloid lineage.20 Both TNF-Rs have been found to inhibit the bioactivity of TNF-α in vitro,21 although there is a report suggesting that binding of TNF-α to its soluble receptors stabilises the trimeric structure of TNF-α and therefore may prolong its bioactivity in vivo.22 It has become increasingly apparent that cytokines do not work in isolation but function as part of a tightly regulated cytokine network in which soluble receptors play an integral part. Thus conditions priming for production of TNF-α would lead to subsequent induction of TNF-R production as part of the normal regulatory feedback mechanism.23 Increased levels of TNF-Rs have been reported in the plasma and the bronchoalveolar lavage (BAL) fluid of subjects with sarcoidosis,24 ,25 but their production in the lung relative to TNF-α bioactivity has not been explored to date.
In this study we have investigated the relationship between TNF-α bioactivity and the presence of TNF-R in the BAL fluid and alveolar macrophage (AM) supernatants of healthy controls and subjects with pulmonary sarcoidosis. The patients were divided on the basis of their chest radiographs into subjects with stage I disease and those with stage II/III disease in order to determine any relationship between TNF-α regulation and the severity of pulmonary involvement.
Methods
SUBJECTS
The subjects with sarcoidosis underwent fibreoptic bronchoscopy with transbronchial biopsy for diagnostic purposes at initial presentation. None of the patients had received corticosteroid therapy. All of the patients were non-smokers and were symptomatic (for example, erythema nodosum, arthralgia, breathlessness) at the time of bronchoscopic examination. Sixteen patients were recruited and staged according to chest radiographic findings (table 1). A diagnosis of sarcoidosis was confirmed by the presence of granulomas on the transbronchial biopsy specimen in all subjects. Thirteen non-smoking healthy controls consented to undergo fibreoptic bronchoscopy with bronchoalveolar lavage. These procedures were approved by the Southmead NHS Trust ethics committee.
Characteristics of study subjects
BRONCHOALVEOLAR LAVAGE
Bronchoalveolar lavage was performed in the right middle lobe. Prior to the procedure subjects were injected intramuscularly with 0.6 mg atropine, followed by intravenous sedation with 0–2 mg alfentanil and 0–10 mg midazolam. Topical lignocaine was administered to anaesthetise the upper airway. Three 60 ml aliquots of phosphate buffered isotonic saline were instilled and gently aspirated into a siliconised bottle kept on ice. The chilled BAL fluid was strained through a single layer of coarse gauze to remove mucus clumps and then spun at 400g for five minutes to recover cells. The resultant cell free fluid was stored at –80°C until analysis. The cell pellet was resuspended in serum-free RPMI 1640 medium (Sigma, Poole, UK) supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma) and adjusted to 1 × 106AMs per ml as defined by morphology.26 The cells were plated out onto sterile 96 well plates (Gibco Nunc) for two hours, 1 × 105 AMs per well. The non-adherent population was then removed by aspiration and 100 μl of fresh RPMI was added to each well. In our laboratory this method has demonstrated >95% pure AMs in the adherent population.26 AMs from 13 healthy controls, six subjects with stage I sarcoidosis, and 10 with stage II/III disease were cultured for 24 hours at 37°C and 5% CO2 in the presence or absence of 10 μg/ml lipopolysaccharide (LPS) (E coli 0111:B4). LPS was used as a non-specific immune stimulus to determine whether the AMs had altered responses in an activated state. Harvested supernatants were stored at –80°C.
MEASUREMENT OF TNF-α BIOACTIVITY
The WEHI 164 clone 13 mouse fibrosarcoma cell line displays dose dependent cytotoxicity in response to TNF-α.27 Confluent cultures were rinsed with sterile phosphate buffered saline, Ca2+ and Mg2+ free, before addition of trypsin:EDTA solution to detach the cells. After centrifugation at 80g for five minutes the cells were resuspended in RPMI 1640 medium (Sigma) containing 20% heat inactivated fetal calf serum (Sigma). 50 μl of the cell suspension was added to the wells of 96 well tissue culture microtitre plates (Gibco-Nunc, Paisley, UK)) and the cells were left to adhere for two hours in an incubator. Following cell attachment, 10 μl of 10 μg/ml actinomycin D (Sigma) was added to each well; 40 μl of TNF-α standards (a gift from Bayer, UK, range 1–500 pg/ml) and samples were added in triplicate to bring the final volume in the well to 100 μl. WEHI cultures were then incubated for 20 hours at 37°C, 5% CO2 in a humidified incubator. Following this, 25 μl of 5 mg/ml 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT; Sigma) was added to each well and incubated for a further two hours. MTT dye is metabolised by viable cells to give a purple formazan product. The cells were then lysed with 100 μl of 20% sodium dodecyl sulphate/50% dimethyl formamide acidified to pH 4.7 with acetic acid. The plates were incubated overnight to dissolve the formazan before reading at 570 nm on a plate reader (Dynatech MR7000). Sample values were extrapolated from the standard curve using the Biolinx package (Dynatech, Billingshurst, UK). This bioassay has a detection limit of 0.003 ng/ml for exogenous TNF-α.
MEASUREMENT OF TOTAL (BOUND AND FREE) TNF-α
Immunoreactive TNF-α was measured by an ELISA which was performed according to manufacturers’ instructions (Quantikine R & D Systems, Abingdon, Oxon, UK; detection limit 0.004 ng/ml). The addition of both p55 and p75 has been proved by the manufacturers to have no effect on the measurement of TNF-α in the assay, and it is therefore recognised that this assay does not discriminate between free and bound TNF-α.
TNF RECEPTOR ELISA
ELISA plates (Nunc Maxisorp) were coated with 5 μg/ml 5R13 monoclonal antibody for p55 plates and 5 μg/ml 7R10 monoclonal antibody for p75 plates (courtesy of Dr Sue Stephens, Celltech, UK) in coating buffer 100 μl per well and incubated for 12 hours at 4°C. These antibodies showed no cross reactivity with other known cytokines and their activity was not affected in the presence of human plasma or serum. The plates were then blocked with PBS containing 0.5% BSA for one hour at 37°C. The wells were washed three times on a plate washer with PBS containing 0.1% Tween 20 (Sigma). Samples were then added, together with the recombinant p55 or p75, 100 μl per well, and the plates were incubated for two hours at 37°C. The plates were washed four times in wash buffer and 100 μl of biotinylated TNF-α (50 ng/ml) was added to each well. The use of biotinylated TNF as a detector restricts this ELISA to the detection of free rather than bound receptors. The plates were incubated for one hour at 37°C and then washed four times with wash buffer. The bound TNF was detected with streptavidin peroxidase 1:400 dilution, incubated for 30 minutes at room temperature, and the plates washed four times. TMB substrate was then added (200 μl per well) and the plates left to develop in the dark for 15 minutes. The reaction was stopped with 50 μl of 1 M sulphuric acid and the plates were read at 450 nm (reference filter 630 nm) on a plate reader. Sample values were calculated from the resultant standard curve. The detection limit was 0.03 ng/ml for exogenous p55 and 0.07 ng/ml for exogenous p75.
DATA ANALYSIS
The data are presented as median values with 95% confidence intervals (CI). The data, which were not normally distributed as determined by the Ryan Joiner normality test, were compared using the Mann-Whitney analysis on Minitab for Windows, a p value of <0.05 being regarded as significant.
Results
BIOACTIVE TNF-α AND TNF-R DETECTED IN THE BAL FLUID
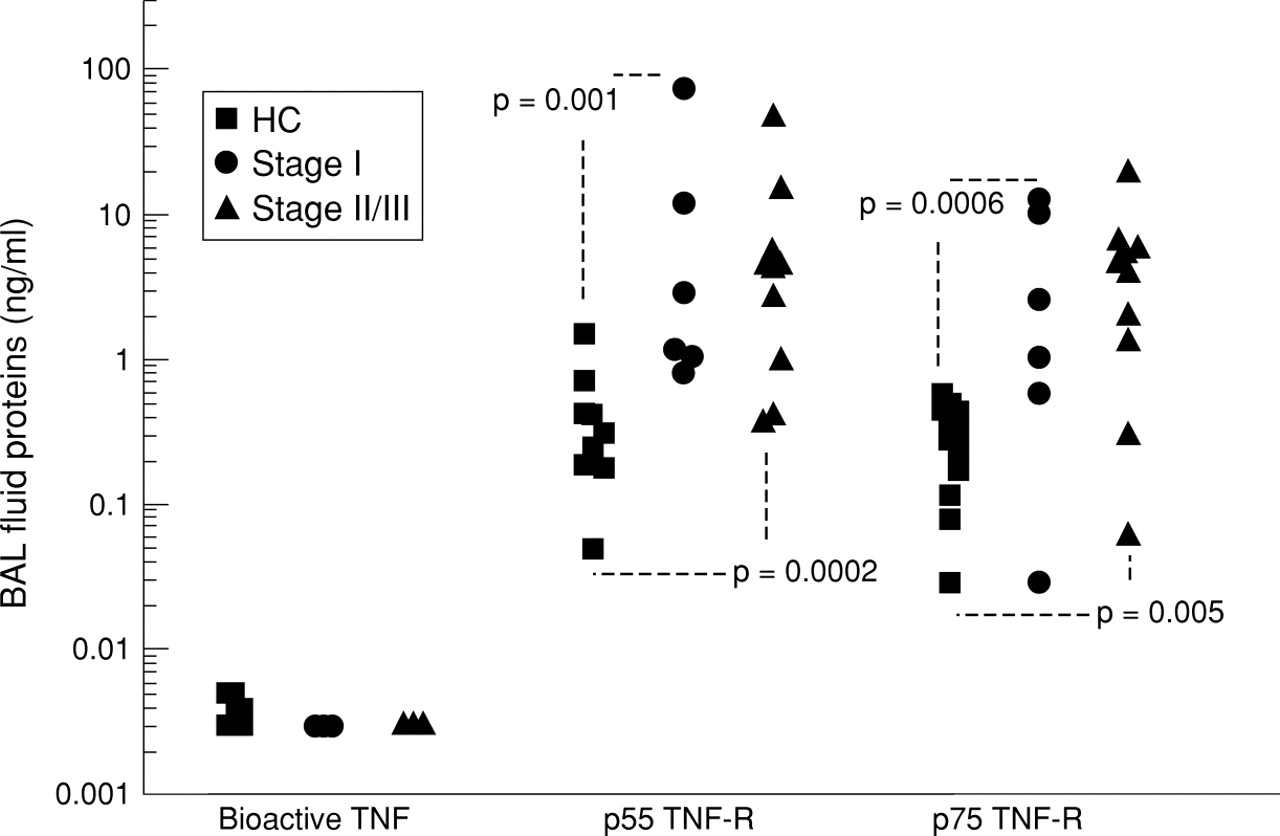
All BAL fluid samples from both groups of subjects (stage I and stage II/III) had bioactive TNF-α levels below the 0.003 ng/ml detection limit for this assay. The median value for normal controls was 0.004 ng/ml (fig 1). The p55 levels were significantly increased in the BAL fluid of patients with stage I disease compared with control subjects (median 2.10 ng/ml vs 0.23 ng/ml, 95% CI 0.79 to 12.31, p = 0.001). This was also true for those with stage II/III disease (median value 4.36, 95% CI 0.81 to 5.25, p = 0.0002). There was no significant difference between the two sarcoidosis groups. For p75 the values were 0.331 ng/ml, 4.629 ng/ml, and 4.371 ng/ml for healthy controls and subjects with stage I and stage II/III disease, respectively. There was no significant difference between the two sarcoidosis groups but the BAL fluid from both those with stage I disease (95% CI 0.648 to 10.416, p = 0.0006) and stage II/III disease (95% CI 1.081 to 5.586, p = 0.005) contained more p75 than BAL fluid from healthy controls.

Bioactive ΤΝF-α and p55 and p75 TNF-R detected in the BAL fluid collected from subjects with stage I (n = 6) and stage II/III sarcoidosis (n = 10) and healthy controls (HC; n = 13).
BIOACTIVE TNF-α DETECTED IN AM SUPERNATANTS
TNF-α release by AMs was suppressed in subjects with stage I sarcoidosis compared with the other groups. The unstimulated values were 0.532 ng/ml, 0.173 ng/ml, and 0.468 ng/ml for controls, patients with stage I sarcoidosis and stage II/III disease, respectively, and were not significantly different. However, the median LPS induced TNF-α in controls was 2.385 ng/ml compared with 0.333 ng/ml in the group with stage I disease (95% CI 0.498 to 2.693, p = 0.004) and 1.362 ng/ml in those with stage II/III disease (fig 2). The difference in LPS induced bioactive TNF-α between the two sarcoidosis groups was also significantly different (95% CI 0.327 to 6.298, p = 0.008). The four highest LPS induced TNF-α levels were observed in cultures derived from patients with stage II sarcoidosis.

Bioactive ΤΝF-α detected by WEHI bioassay in untreated and LPS treated AMs derived from subjects with stage I (n = 6) and stage II/III sarcoidosis (n = 10) and healthy controls (HC; n =13).
P55 TNF-R DETECTED IN AM SUPERNATANTS
p55 release by AMs derived from subjects with stage I sarcoidosis was increased compared with the other groups. In unstimulated control cultures the median p55 was 0.088 ng/ml compared with 0.476 ng/ml in the group with stage I sarcoidosis (95% CI 0.161 to 0.762, p = 0.0008) and 0.033 ng/ml in those with stage II/III disease (fig 3). p55 release by unstimulated AMs was significantly higher in the stage I group than the stage II/III group (95% CI 0.132 to 0.756, p = 0.008). There were also similar differences observed in LPS stimulated cultures between control subjects and those with stage I disease (medians 0.078 ng/ml and 0.449 ng/ml, respectively, 95% CI 0.081 to 0.07, p = 0.003) and between those with stage I and stage II/III disease (median stage II/III disease 0.058 ng/ml, 95% CI 0.069 to 0.530, p = 0.01).

p55 TNF-R detected by ELISA in untreated and LPS treated AMs derived from subjects with stage I (n = 6) and stage II/III sarcoidosis (n = 10) and healthy controls (HC; n =13).
P75 TNF-R DETECTED IN AM SUPERNATANTS
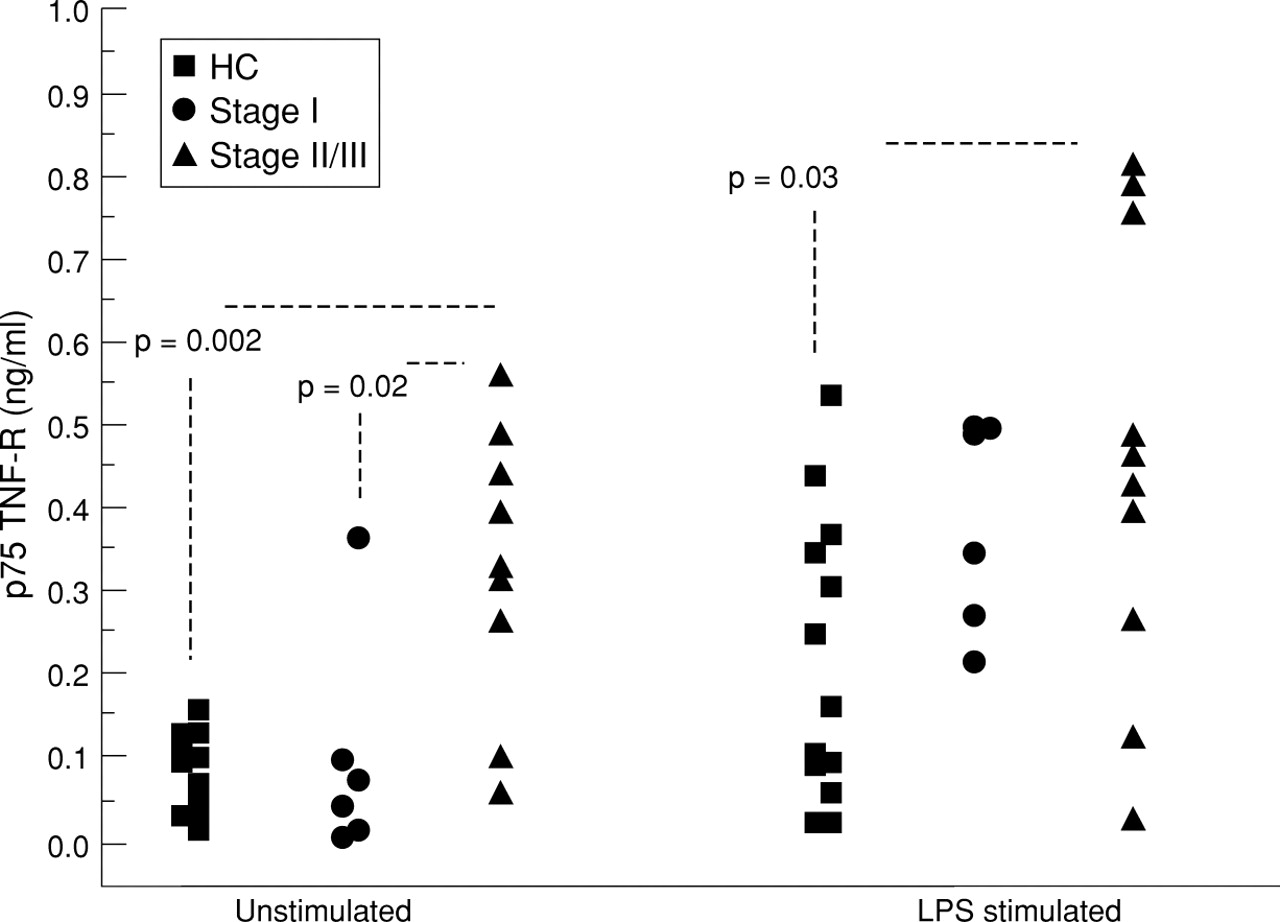
In unstimulated cultures the subjects with stage II/III disease released more p75 than those with stage I disease and healthy controls (median values 0.326 ng/ml vs 0.064 ng/ml (95% CI 0.028 to 0.395, p = 0.02) and 0.102 ng/ml (95% CI 0.157 to 0.362, p = 0.002) respectively; fig 4). When AMs were stimulated with LPS the median values were 0.448 ng/ml, 0.424 ng/ml, and 0.169 ng/ml, respectively, for subjects with stage II/III disease, stage I disease, and controls. Only the difference between controls and those with stage II/III disease remained significant (95% CI 0.02 to 0.437, p = 0.03).

p75 TNF-R detected by ELISA in untreated and LPS treated AMs derived from subjects with stage I (n = 6) and stage II/III sarcoidosis (n = 10) and healthy controls (HC; n =13).
DETECTION OF IMMUNOREACTIVE (BOUND AND FREE) TNF-α IN COMPARISON TO BIOACTIVE TNF-α IN BAL FLUID AND AM SUPERNATANTS
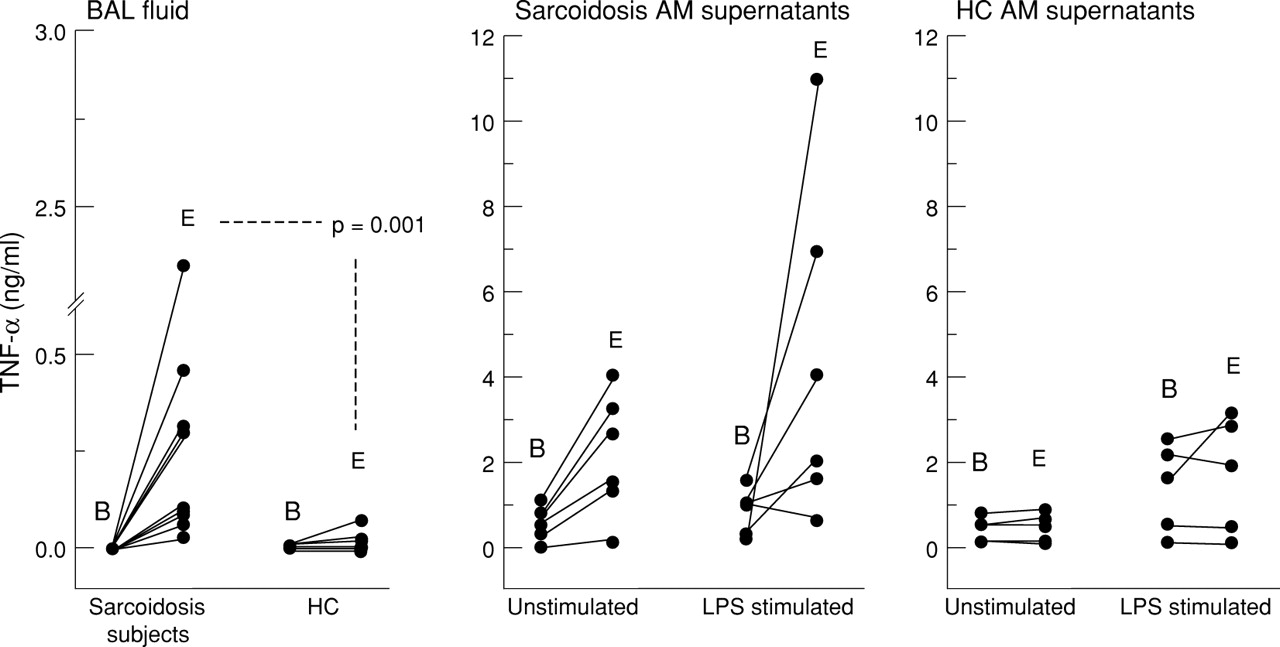
A comparison between immunoreactive and bioactive TNF-α was made on BAL fluid derived from all subjects studied. Matched AM supernatants were derived from only five healthy controls and six subjects with sarcoidosis (two stage I and four stage II/III) due to scarcity of supernatant. The detection of immunoreactive TNF-α was increased compared with bioactive TNF in most samples from both subject groups, but the increase was greatest overall in the sarcoidosis population (fig 5). There was increased detection of immunoreactive TNF-α to bioactive TNF-α in the BAL fluid derived from subjects with sarcoidosis. There was also significantly increased total TNF-α in the BAL fluid derived from subjects with sarcoidosis compared with healthy controls (median values 0.304 ng/ml and 0.004 ng/ml, respectively, 95% CI 0.076 to 0.455, p = 0.001). Although there was increased detection of immunoreactive TNF-α compared with bioactive TNF-α in AM supernatants from both groups, no statistically significant differences were observed. The two samples with the highest detectable TNF-α in both unstimulated and LPS stimulated cultures were derived from subjects with stage I sarcoidosis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Detection of bioactive TNF-α and immunoreactive (bound and free ) TNF-α as determined by WEHI bioassay (B) and R & D Quantikine ELISA (E), respectively. BAL fluid from both subject groups (n = 10), AM supernatants from both subjects with sarcoidosis (n = 6) and healthy controls (HC; n = 5).
Discussion
This is the first study to compare TNF-α bioactivity, the presence of soluble TNF receptors, and total TNF-α in subjects with stage I and stage II/III sarcoidosis versus healthy controls. We did not detect a significant increase in TNF-α bioactivity in either the BAL fluid or AM supernatants derived from subjects with stage I or stage II/III sarcoidosis compared with control subjects, although the highest bioactive TNF-α levels were detected in the stage II/III population. The stage I population actually had significantly reduced TNF-α bioactivity compared with the other two groups. The role of TNF-α in the pathogenesis of sarcoidosis remains controversial, with variable findings having been reported in studies of BAL fluid and AM culture supernatants,12-16 although the majority suggest increased TNF-α production. Most of the previous reports relied on radioimmunoassay or ELISA to determine TNF-α levels, which did not discriminate between bound and unbound cytokine. Some reports have referred to a correlation between the L-cell bioassay and the ELISA they employed but have provided no data in support of this.13 ,14 By using the R & D ELISA (which determines total TNF-α levels) alongside the WEHI bioassay we have shown that, in the lungs of our sarcoidosis subjects, much of the TNF-α is in the biologically inactive form. In a previous study Bachwich and co-workers used an alternative bioassay (using the L929 cell line) to measure TNF-α in subjects with active and inactive sarcoidosis.16 They also found no increase in TNF-α bioactivity in the BAL fluid or AM supernatants, with the exception of AMs from active sarcoidosis subjects stimulated with 10 μg/ml LPS in whom a small but significant increase was reported. We observed a similar response in our stage II/III population. The fact that the results of our study and those of Bachwich et al are largely comparable using similar methods highlights the importance of using bioassays whenever possible when implicating a cytokine in a disease process.
In this study we have made the distinction between stage I disease, which often has a self-limiting clinical course requiring no treatment, and stage II and III disease where there may be progressive lung involvement and impairment of lung function. Our findings suggest that TNF-α bioactivity is restricted in subjects with stage I disease, and this may be an important factor which prevents the development of interstitial changes despite the presence of a mononuclear cell alveolitis and granulomas.
A likely explanation for reduced bioactivity in stage I disease is the inhibitory activity of soluble TNF-R, especially p55. We have found significantly increased levels of p55 and p75 TNF-R in the BAL fluid derived from subjects with stage I and stage II/III disease compared with healthy controls, and enhanced production of p55 and p75 TNF-R by stage I and stage II/III AMs, respectively, in vitro. The presence of increased TNF-R in these biological fluids cannot be explained by less binding to reduced amounts of free TNF-α since detection of free and bound TNF-α in the BAL fluid using the R & D ELISA kit was significantly increased in the sarcoidosis group. The ability of soluble TNF-Rs to neutralise the biological effects of TNF-α has been highlighted by in vivo studies. p55 TNF-R infusion confers resistance to the lethal effects of TNF-α28 and mice transgenic for p55 TNF-R demonstrate resistance to LPS infusion.29 Our data suggest that, in subjects with stage I disease, there may be a shift in the balance of TNF/TNF-R in favour of inhibition, an observation reported in normal AMs both in this and in another recent study.30 This is also supported by a study which found significantly increased levels of p75 in the BAL fluid and a significant correlation between p75 levels and lymphocyte numbers in patients with sarcoidosis.25 The autocrine regulatory mechanism we propose for AMs is similar to that observed for monocytes from normal blood,31 a finding of interest since it has been shown that AMs in the lungs of subjects with sarcoidosis bear an immature phenotype which closely resembles blood monocytes. This suggests that they may be newly recruited from the systemic circulation.32
It is widely accepted that TNF-Rs are successful inhibitors of TNF-α bioactivity in vivo28 ,33 and that they are part of the regulatory response in acute diseases such as meningitis34and sepsis35 ,36 as well as in response to LPS challenge in experimental conditions.37 However, increased levels of TNF-R have been found to correlate with mortality in one study on patients with sepsis.37 Infusion of low levels of TNF-R has resulted in an increased plasma half life for TNF-α in animal models of endotoxinaemia, which suggests that their presence can be associated with increased injury rather than protection.31This implies that limiting ΤΝF-α bioactivity in the acute inflammatory response may lead to slow resolution and possibly chronicity. Since the aetiology of sarcoidosis is presently unknown, this theory cannot be applied directly but it does demonstrate how so called beneficial mediators could be deleterious in certain circumstances.
In summary, TNF-α bioactivity is reduced in the lungs of subjects with stage I sarcoidosis compared with subjects with stage II/III disease and healthy controls. This reduced bioactivity may be a consequence of increased TNF-R production. Whilst it must be appreciated that BAL fluid and BAL derived AMs do not necessarily reflect the inflammatory processes within the sarcoid granuloma itself, this report does provide evidence for an important homeostatic mechanism which may prevent the development of interstitial lung changes more characteristic of stage II/III sarcoidosis.
Acknowledgments
Dr Lynne Armstrong is funded by The Sir Jules Thorn Charitable Trust.