Article Text

Abstract
The diagnosis of cystic fibrosis (CF) is based on the occurrence of two mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and on assays that measure the basic defect of abnormal chloride transport in the affected organs. However, in cases of atypical CF not all diagnostic tests may be positive. We present a patient with an atypical CF phenotype in whom the only presenting symptom was severe CF-like lung disease substantiated by an abnormal nasal potential difference. Genetic analysis showed that the patient was a symptomatic heterozygote, which suggests that one lesion in theCFTR gene may be sufficient to cause CF-like lung disease.
- cystic fibrosis
- atypical cystic fibrosis
- nasal potential difference
- genetics
Statistics from Altmetric.com
Typical cystic fibrosis (CF) is caused by two lesions in the cystic fibrosis transmembrane conductance regulator (CFTR) gene which give rise to a generalised exocrine disease of the respiratory, gastrointestinal, reproductive, and hepatobiliary tracts.1 The protein product of theCFTR gene is a chloride channel expressed in the apical membrane of epithelial cells2 so diagnostic tests that measure the chloride conductance in exocrine epithelia—for example, the pilocarpine iontophoresis sweat test,3intestinal current measurement (ICM),4 and nasal potential difference (NPD)5 ,6—have abnormal values in cases of typical CF. Atypical cases of CF have a different clinical presentation with pancreatic sufficiency, mild bronchitis, nasal polyposis, congenital bilateral absence of the vas deferens (CBAVD), a borderline sweat test, or ICM values which point to low residual chloride secretion in intestinal tissue either by CFTR or an alternative chloride channel.7-10 We present a case characterised exclusively by severe lung disease in whom other organs typically involved in CF were not affected and who failed to show a defect in chloride transport.
Patient history
The 34 year old patient was the third child of Caucasian first generation cousins. The first child was born preterm with meconium ileus and died at day 10. The second child died during infancy at six months and CF was suspected at necropsy. Our patient suffered from chronic nasal polyposis which had led to 13 polypectomies between the age of five and 23 years. By the age of 15 years she was referred to a chest physician because of shortness of breath during sports activity. Clubbing, subnormal lung function, and decreased exercise tolerance were noted. The diagnosis of CF was proposed because of the typical pulmonary manifestations but was discarded when normal sweat electrolyte concentrations were found. The differential diagnosis of allergy as the underlying disease was excluded by normal IgE skin prick tests and normal serum levels of specific IgE. Immotile cilia syndrome was excluded by the normal microscopic appearance of biopsy specimens of nasal cilia. Since adolescence she had been underweight (below 3rd percentile) and had recurrent lower airway infections. Over the years increased sputum production and chronic cough became a clinical problem. A lung biopsy specimen in 1984 showed a histological pattern consistent with CF (bronchiectasis with localised purulent bronchitis and surrounding fibrosis). Cultures of throat swabs or sputum were often positive for Staphylococcus aureus(>90% of samples) but never forPseudomonas aeruginosa. However, the specific anti-P aeruginosa oprF IgG titre was positive, indicating that she had been exposed to P aeruginosa. Bronchodilators and intermittent antibiotics were prescribed but the patient generally discontinued medication after a few days, even during acute respiratory tract infections. At the age of 23 her lung function was reduced (vital capacity 1.5 l (48% predicted)), her chest radiograph had a Chrispin-Norman score of 20, grade 3 (range 0–38), her height was at the 25th percentile and weight below the 3rd percentile. However, her pancreatic function was sufficient as shown by normal chymotrypsin levels in 1986, 1992, 1995, normal stool elastase levels (386 mg/g) in 1995, and by ultrasound investigation of the pancreas. She had never received pancreatic enzymes or vitamin supplements. Serum levels of vitamins (vitamin A 370 μg/l, vitamin E 11 mg/l), bilirubin and liver enzymes were always in the normal range. Moreover, in repetitive pilocarpine iontophoresis sweat tests performed since she was 11 years old the sweat electrolyte levels were always normal (range 7–32 mEq/l).
Methods
This study was approved by the medical ethical committees of the Dutch and German participating hospitals and informed consent was obtained from the patient. All chemicals were obtained from the Sigma Chemical Co, St Louis, Missouri, USA.
ANALYSIS OF THE CFTR GENE
The promoter (–3.8 to –0.3 kb) and coding regions and the exon flanking intron sequences of the CFTRgene were screened for mutations by single strand conformation polymorphism (SSCP) analysis11 ,12 and, in the case of the appearance of non-wild type band patterns in the high resolution SSCP gel, by subsequent sequencing.13 Deletions in theCFTR gene were sought by repeated Southern hybridisations of macrorestriction blots with CFTR cDNA probes encoding exon 1, exons 7–24, or the second nucleotide binding fold (codons 1202–1422). Preparation of unsheared genomic DNA from fresh blood, complete restriction digestions with ApaI,EagI, FspI,SalI or XhoI, pulsed field gel electrophoresis, blotting and hybridisation followed protocols 1, 5, 7, 8 and 9 of Bautsch et al.14 The intragenicCFTR haplotype was determined for the sequence alterations T854T and M470V,15 ,16 the splice site polymorphism in intron 8 (TG)mTn,11 ,17 and the microsatellites IVS8CA, IVS17bCA, IVS17bTA.18 ,19
NASAL POTENTIAL DIFFERENCE (NPD)
NPD was measured using an adaptation of the method described previously.5 In short, the nasal turbinate was superfused (1.7 ml/min) for periods of three minutes with the following solutions (mol/l): saline to measure baseline PD; amiloride (10–4) to block Na+ channels; gluconate with amiloride to assess spontaneous Cl– conductance; isoprenaline (10–4) in gluconate with amiloride to evaluate the presence of CFTR, and ATP (10–3) in isoprenaline/gluconate+amiloride solution to open non-CFTR Cl– channels. To access the submucosal space a small needle (25 gauge) was inserted into the forearm and filled with saline. Both the needle and superfusion catheter were connected to a high input resistance voltage measuring device via salt bridges and Ag/AgCl electrodes. The saline solution consisted of (mmol/l): NaCl (120), Na gluconate (25), K gluconate (5), NaH2PO4 (0.4), Na2HPO4 (2.4). In the gluconate solution NaCl was replaced by Na gluconate (145). All solutions were adjusted to pH 7.4.
INTESTINAL CURRENT MEASUREMENT (ICM)
The method used to study ICM has been described previously.4 Freshly obtained rectal biopsy specimens were mounted in saline solution in the Ussing chamber (aperture 1.2 mm). After stabilising the basal short circuit current (Isc) the tissue was exposed to the following inhibitors and secretagogues (mol/l) to the mucosal (M) or serosal (S) side: glucose (10–2, M+S); amiloride (10–4, M); indomethacin (10–5, M+S) to inhibit basal Cl–secretion by inhibiting the endogenous prostaglandin formation; carbachol (10–4, S), a Ca2+ linked secretagogue; forskolin (10–5, M+S) + 8-bromo cyclic adenosine monophosphate (cAMP, 10–3, M+S) to open Cl– channels by activation of a cAMP-dependent protein kinase and phosphorylation of specific membrane proteins; 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS, 2.10–4, M) to inhibit Ca2+ dependent Cl– secretion; and histamine (5.10–4, S) to reactivate the Ca2+ activated pathway.
Results
CFTR GENETICS
Despite the familial consanguinity, the patient was heterozygous at the CFTR locus which is shown by the different number of (TA)n repeats (n = 30, 34) for the microsatellite at the locus IVS17bTA and her heterozygosity for a rareCFTR mutation. She was homozygous (TG)12T7-2-1 for theCFTR haplotype (TG)mTn-M470V-T854T. The combination of the TG12 repeat with the T7 allele and the V470 CFTR isoform are known to decrease significantly the amount and chloride channel activity of CFTR.16 After allCFTR exons and flanking intron sequences and most of the promoter area up to –4 kb had been screened by SSCP, one molecular lesion (the splice site consensus transition 1898+3 A->G) was identified. A pancreatic insufficient child with CF at our clinic is homozygous for this condition and exhibited the typical pulmonary and gastrointestinal manifestations of CF indicating that 1898+3 A->G is a CF-causing lesion. No anomalous bands were seen in autoradiograms of macrorestriction blots probed with CFTR cDNAs, indicating that the two CFTR alleles of our patient do not carry any major genomic alteration (fig1).

Autoradiogram of genomic ApaI digestions probed with CFTR cDNA (exon 7–24). No anomalous band pattern is seen for the sample from the patient (lane 1). For comparison, lane 4 shows the probe reactive fragments from a specimen which carries a deletion in one CFTR allele. ApaI cleaved fragments were separated in a CHEF-DRTMII cell at 5.6 V/cm in 1% agarose gels (0.5 TBE buffer, 10°C). Pulse times were linearly increased in two ramps from 5 to 20 s in 18 hours and from 5 to 90 s in 20 hours.
DIAGNOSTIC ASSESSMENT
On the day of electrophysiological investigation at the age of 32 she was underweight (below the 3rd percentile with a body mass index of 17 kg/m2) and her lung function was severely reduced (FEV1 0.39 l (14% predicted), FVC 1.23 l (38% predicted)). At this time she was treated with continuous oxygen and showed severe clubbing. Electrophysiological measurements were carried out by assessing the presence of chloride conductance in her airway and intestinal epithelium (table 1). The basal NPD was –52 mV (fig 2). In the presence of amiloride the PD depolarised to –31 mV, a decrease of 40%. Superfusion of gluconate in the presence of amiloride resulted in a net response of –3 mV, indicating subnormal Cl–conductance. With isoprenaline, which opens CFTR Cl–channels, only a small response of –4 mV was obtained which suggested that few CFTR channels are present. No ATP response, which is usually indicative of the presence of alternative Cl–channels,20 was seen in the airway tissue.
Electrophysiological results for the study patient. Mean (SD) values for a control group and a group of patients with CF are given for comparison
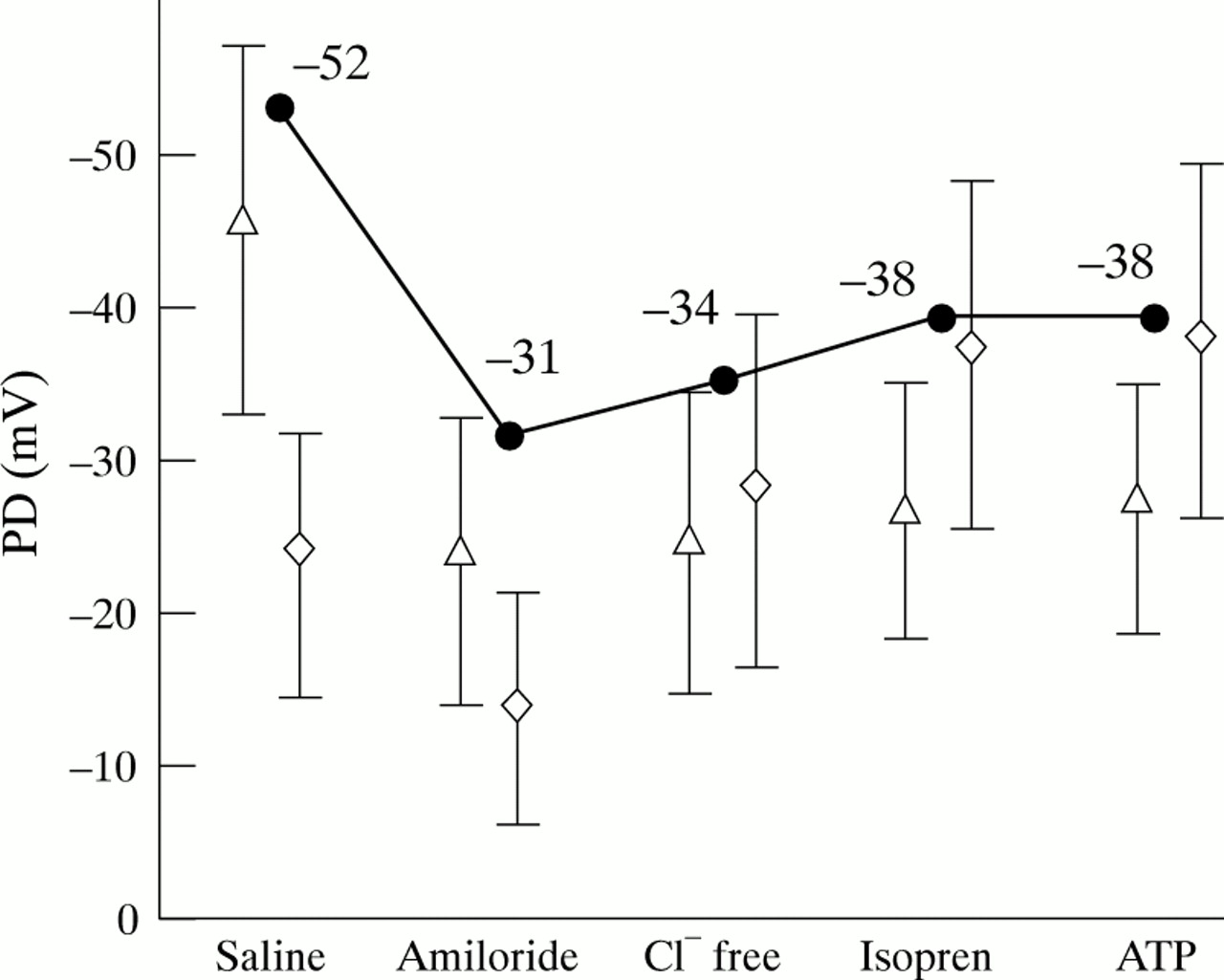
Nasal potential difference (PD) measurements of the patient (•) and mean (SD) PD values of 25 controls (⋄) and 23 patients with CF (Δ) following superfusion with saline solution, amiloride (10–4 M) in saline solution, Cl–free solution with amiloride, isoprenaline (10–4 M) in Cl– free solution with amiloride, and ATP (10–3 M) in Cl– free solution with amiloride and isoprenaline.
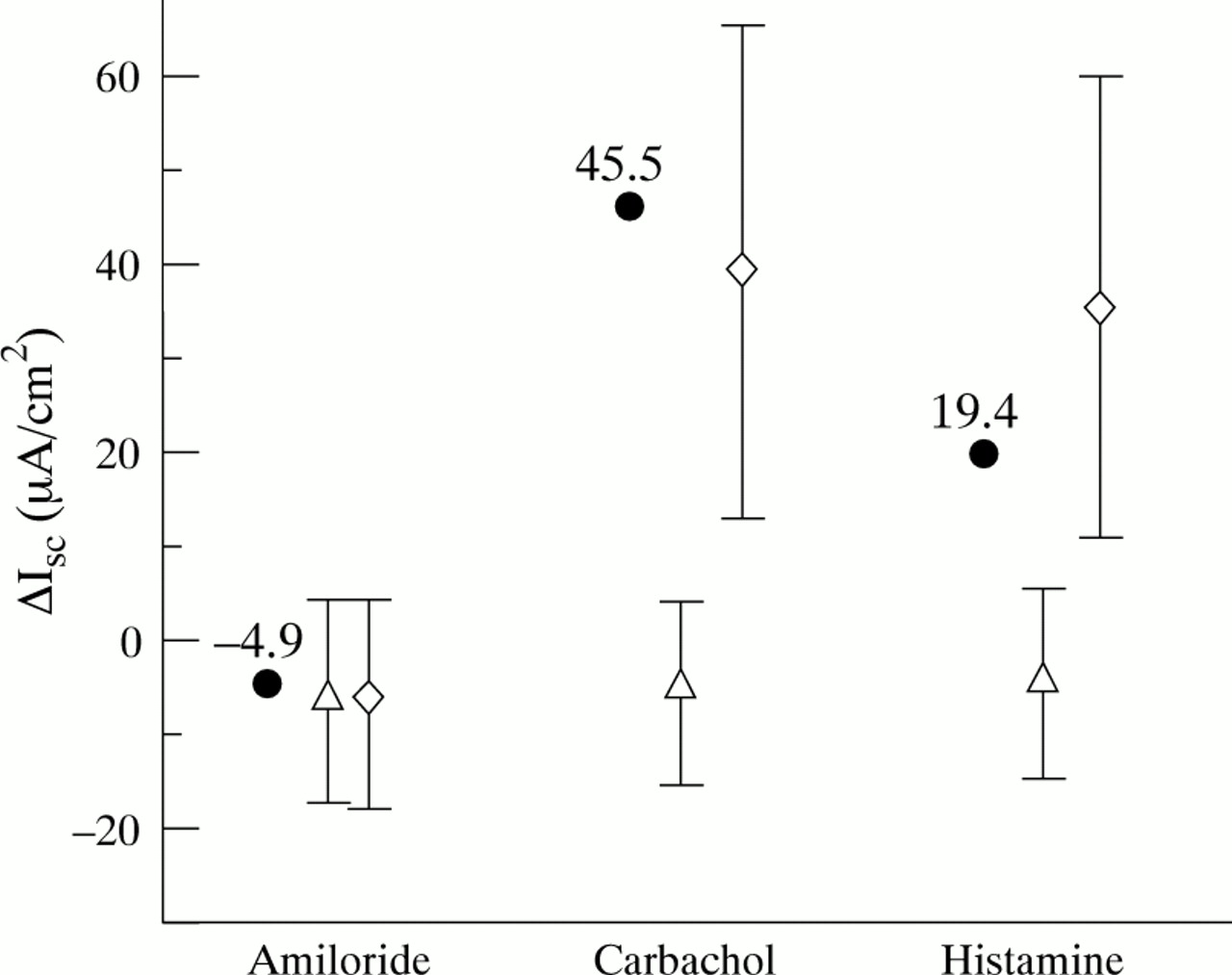
In previous ICM studies carbachol has provoked a negative reversed Isc in rectal tissue of patients with CF, sometimes followed by a positive residual response indicating the presence of residual Cl– secretion. In our patient the ICM showed a Cl– secretory current in the normal range on addition of carbachol (fig 3). DIDS, which inhibits the alternative pathway of Ca2+ activated Cl– currents, did not influence the Cl– current, suggesting the presence of normal CFTR mediated chloride conductance in the intestine.

{kind=link}
{kind=link}
{kind=link}
Measurements of intestinal current expressed as changes in short circuit current (Isc) of the patient (•) and mean (SD) Isc of 50 controls (⋄) and 51 patients with CF (Δ) following the addition of amiloride, carbachol, and exposure to 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS) + histamine.
Discussion
The clinical and diagnostic features presented by this case are conflicting in terms of the expression of the basic defect in different tissues. In typical CF defective electrolyte transport in the sweat gland, intestinal and airway epithelium is shown by a pathological outcome of the sweat test, ICM and NPD. In our case, however, the sweat chloride concentration and ion flow in the intestine were normal while the NPD values were abnormally high. Pathologically raised NPD values have so far only been found in CF and not in any other lung disease with related clinical manifestations such as α1-antitrypsin deficiency, immotile cilia syndrome, or congenital bronchiectasis.21 As well as a high baseline nasal PD, the patient’s gluconate and isoprenaline response were also in the CF range. However, they do point to the presence of little residual CFTR Cl– permeability in her respiratory epithelium. The clinical symptoms of our patient, such as progressive obstructive bronchitis and bronchiectasis, sputum production, and pathological bacterial flora, are typical of CF and she has been permanently on oxygen since the age of 30. This could explain her anorexic status since there is no sign of malabsorption or gastrointestinal disease, as confirmed by normal vitamin A and E levels, pancreatic sufficiency, and a normal ICM. This highly discordant expression of the basic defect in different tissues is described here for the first time: CFTR function was unaffected in gastrointestinal tissue whereas in airway tissue the abnormally low Cl– conductance can only be accounted for by defective epithelial Cl– transport. Other cases of CF with normal sweat test results and pulmonary disease, as described for patients carrying the A455E or 3849+10 kb C->T mutation,7 ,8 ,22 ,23 can clearly be diagnosed by an abnormal ICM even when there are few or no clinical signs of gastrointestinal involvement.10
The CFTR gene was screened for disease causing lesions in all exons and flanking intron sequences and on one chromosome a sequence alteration in a donor splice site was found (1898+3 A->G). This sequence alteration was first described in a compound heterozygous pancreatic sufficient patient (Ferrariet al, personal communication) and was found also in a pancreatic insufficient CF patient homozygous for this condition (see above). According to CFTRmutation analysis and the family anamnesis, our patient is a carrier for one CFTR mutation and hence should not express any CF symptoms. However, she carries an unfavourable combination of common intragenic polymorphisms. She is homozygous TG12T7 for the TGmTnpolymorphism at the intron 8 splice acceptor site, and the TG12 repeat together with the T7 stretch places the branch point nucleotide in an unfavourable position for splicing leading to 30% of exon 9− CFTR transcript which are known to be translated in CFTR proteins that do not mature.16 ,24 Moreover, she is homozygous for the V470 allele in exon 10. V470 CFTR proteins have a 1.7 fold decreased intrinsic chloride channel activity compared with M470 CFTR proteins.16 These predisposing polymorphisms in the intragenic background decrease the expression and function of CFTR. Yet, they are present in both rectal and nasal tissue and cannot account for the unimpaired function of CFTR in the intestine while in the nasal tissue almost complete absence of Cl–conductance was found. However, the patient’s consanguineous descent leads to a genome-wide overrepresentation of homozygous genotypes which may lead to further unfavourable combinations of factors that modify CFTR expression or lung function. As gastrointestinal disease is not present, we propose that the genetic background predisposes to severe lung disease caused by tissue specific regulatory elements which lead to the loss of CFTR function exclusively in the respiratory epithelium.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft.