Article Text

Statistics from Altmetric.com
Lymphangioleiomyomatosis, a rare disease of unknown aetiology, affects women only. It mainly involves the lungs where, as its name suggests, lymphatics (lymph), blood vessels (angio), and airways are surrounded by smooth muscle (leiomyo) proliferation. It is characterised by progressive dyspnoea, haemoptysis, pneumothorax, and chylous pleural effusions and runs a variable course culminating usually in respiratory failure.1 ,2 Lymph nodes in the abdomen and pelvis may be involved and up to half the patients have renal angiomyolipomas. The disease usually arises spontaneously although it also occurs in some patients with tuberous sclerosis.3
Much of our knowledge of this unusual disorder is anecdotal, coming from case reports and reviews of patients attending tertiary referral centres, which may not represent the full spectrum of the disease. This will change over the next few years as patient registries are being compiled for research purposes in the United Kingdom, United States, and France. This approach, coupled with recent advances in the cell biology of lymphangioleiomyomatosis, will it is hoped improve management and the outcome for patients. This article will cover the clinical features and natural history of isolated lymphangioleiomyomatosis, discuss issues in diagnosis and management, and finally review what is known of the aetiology and cell biology of the disease.
Clinical features, diagnosis, and management
INCIDENCE AND DEMOGRAPHIC FEATURES
The reported prevalence of lymphangioleiomyomatosis is around one per million in the United Kingdom,4 France,5and the United States, although the true prevalence is likely to be greater. The disease is well described in Asia6 although data on prevalence are not available.
Lymphangioleiomyomatosis is exclusively confined to women, the mean age of onset being 34 years.1 ,2 ,6-8 Presentation after the menopause is very unusual, occurring in only eight of 186 patients in the larger series,1 ,2 ,4 ,6-8 six of whom were taking oestrogen containing hormone replacement therapy. This fits with evidence suggesting that oestrogen administration9 and pregnancy10-12 may accelerate disease progression whereas oophorectomy and progesterone2 ,4 may reduce it (see below).
PATHOLOGICAL FEATURES
The lung in lymphangioleiomyomatosis contains numerous cysts, ranging in diameter from millimetres to centimetres,1 and these are responsible for the pneumothoraces and the striking appearance of blebs over the lung surface (fig 1). Infiltration of lung and pleural lymphatics causes obliteration and dilatation of the lymphatic channels leading to lymph stasis, chyloptysis, and septal lines on the chest radiograph. The thoracic duct is often thickened and formed of multiple narrow channels leading to chylous pleural effusions and ascites. Pulmonary vascular occlusion can cause pulmonary haemorrhage, haemoptysis, and haemosiderosis.1
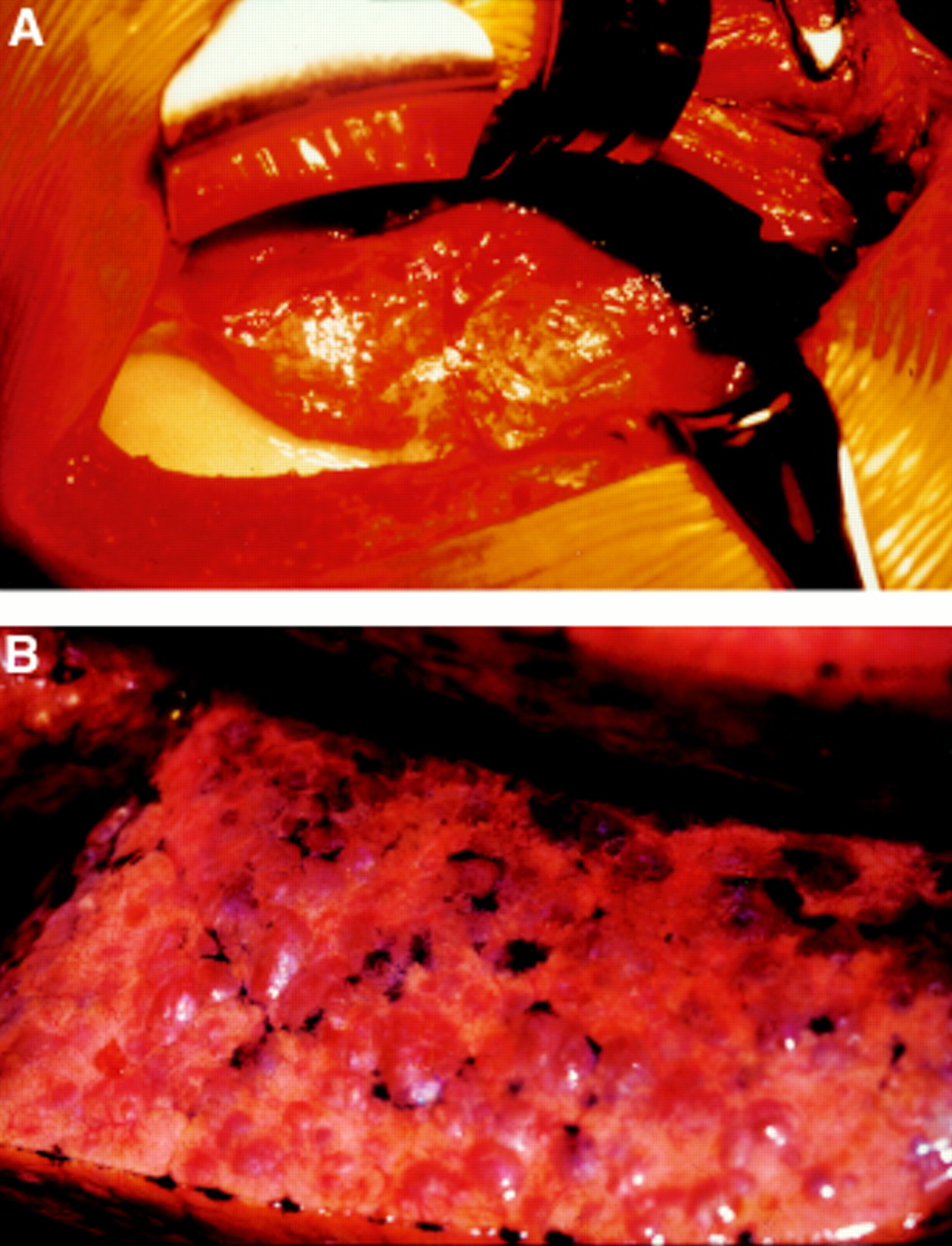
(A) Thoracotomy showing abnormal lung surrounded by a chylous collection. (B) Close up of the lung showing the characteristic appearance of multiple blebs.
Microscopically the lungs are characterised by cystic air spaces and a nodular proliferation of abnormal smooth muscle cells (LAM cells). In early lesions LAM cells accumulate in the alveolar walls, collagen and elastic fibres are partially degraded, and oedema, haemorrhage and haemosiderin laden macrophages are seen around the alveoli.13 ,14 With more advanced disease the nodules are larger and associated with compact collagen bundles whereas oedema, haemorrhage, and macrophages are less prominent.
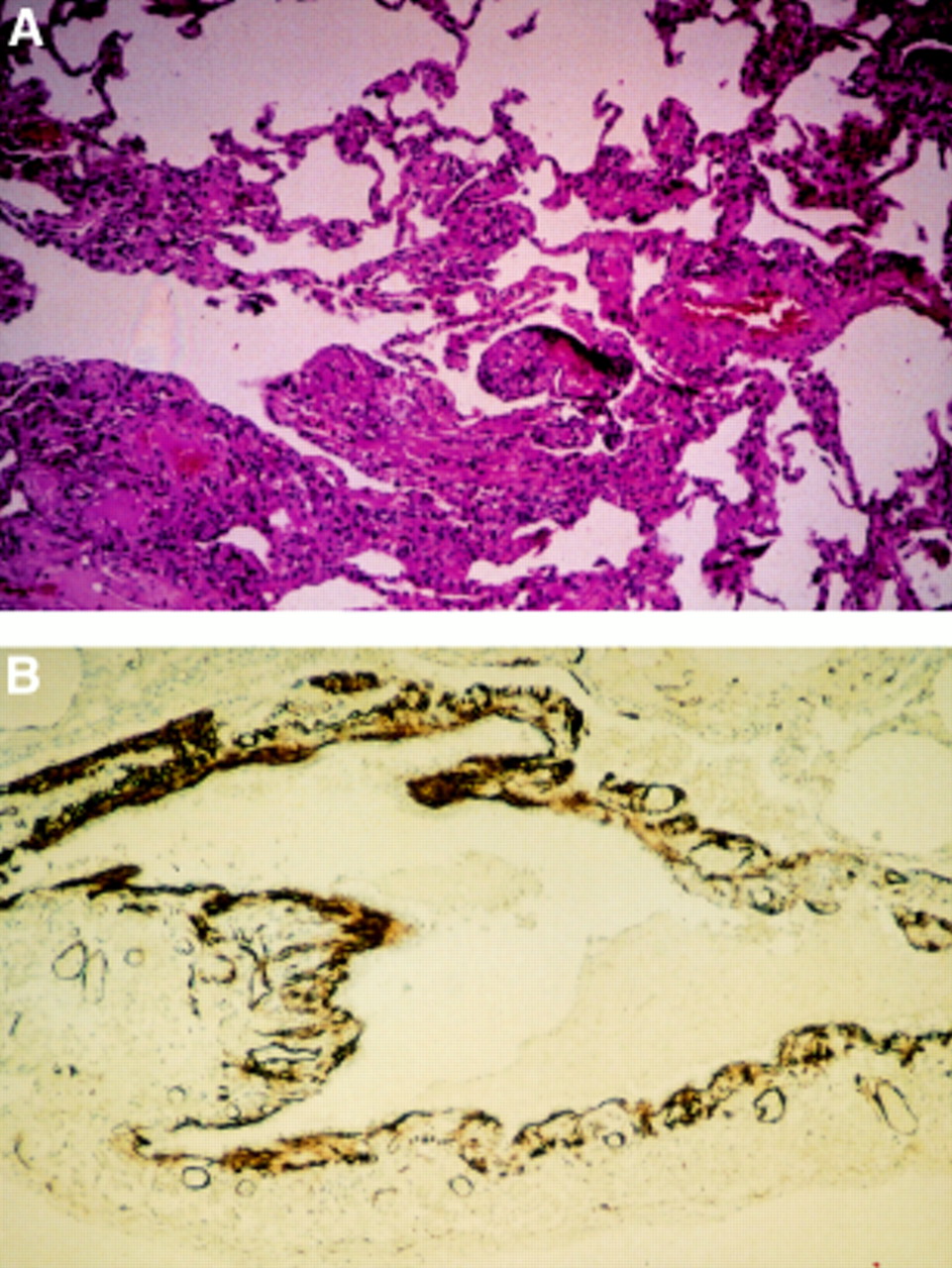
The thin walled cysts are lined by flattened epithelial and ciliated bronchiolar epithelial cells14 and are thought to be formed by the amalgamation of damaged alveoli, probably as a result of uncontrolled proteolytic activity.13 ,14 The nodules surround and may jut into the cysts, alveoli, and bronchioles causing thickening of the small airway walls, often in an irregular fashion (fig 2). LAM cells appear to be of smooth muscle origin since they stain for vimentin, desmin, and α-smooth muscle actin,15-17 and myofilaments are seen on electron microscopy. The cells also display features that are not typical of muscle cells—namely, multiple indented round nuclei, a prominent endoplasmic reticulum, microfilament bundles with dense bodies, numerous electron dense membrane bound granules13 ,18—and they characteristically stain with the monoclonal antibody HMB 45.19 ,20 There appear to be three forms of LAM cell according to their microscopic appearance, large spindle shaped cells, smaller cells with little cytoplasm, and epithelioid cells.14 Interestingly, the three cell types also have differing immunophenotypes which suggests that they have different functions.13 ,14 Receptors for oestrogen and/or progesterone are found on cells from roughly half the patients.21-24 The significance of these findings is uncertain and is discussed below.

Photomicrographs showing nodular proliferation of abnormal smooth muscle and cysts in (A) a lung biopsy specimen stained with haematoxylin and eosin and (B) brown HMB 45 positive LAM cells lining a cystic space.
Angiomyolipomas are rare mesenchymal tumours containing smooth muscle, fat and blood vessels, which also contain HMB 45 positive smooth muscle cells.
CLINICAL FEATURES
Patients with lymphangioleiomyomatosis present most commonly with dyspnoea (59%), pneumothorax (49%), and cough (39%) according to the larger series.1 ,2 ,5 ,6 ,25 Chest pain (22%), chylous pleural effusions (13%), haemoptysis (18%), and wheeze are less common. Chyloptysis, presenting as white, sticky sputum, may occur7 and, following pleurodesis, patients may complain of occasional gurgling in their chest (possibly due to ruptured cysts). Extrapulmonary features include lymph node masses, cystic soft tissue masses,26 ,27 and chylous ascites in up to a third of patients.1 ,25 Uterine fibroids (leiomyomas)8and renal angiomyolipomas28 ,29 are also seen; angiomyolipomas are often asymptomatic although bleeding may occur (see below).
The natural history of lymphangioleiomyomatosis is of progressive airflow obstruction leading to respiratory failure and cor pulmonale.1 The rate of progression is highly variable between patients, ranging from a rapid decline over a few years1 to a more indolent disease over two or three decades.2 ,30 In a review of 47 patients with lymphangioleiomyomatosis in the UK the mean fall in forced expiratory volume in one second (FEV1) was 118 ml/year although there was marked variability between patients.4 Two thirds of women will develop a pneumothorax and these were recurrent in more than two thirds of our patients. Chylous pleural effusions occur in a fifth of patients.5
Estimates of life expectancy in lymphangioleiomyomatosis are still relatively uncertain as previous figures were based on series from tertiary referral centres and the advent of CT scanning may enable the diagnosis to be made earlier. The most recent series reports a 10 year survival of 79%5 but some caution is needed since case ascertainment will inevitably favour those with a better prognosis.
Lung function tests in patients with lymphangioleiomyomatosis show a variety of abnormalities with airflow obstruction and impaired gas transfer the predominant features. Restrictive lung defects are sometimes seen but usually in combination with airflow obstruction and often as the result of pleural effusions, pleurectomy, or thoracotomy.1 ,6 ,31 ,32 Lung function is occasionally normal at presentation6 ,32 but disease progression is associated with increased airflow obstruction and impaired gas transfer.25
DIAGNOSIS
Because lymphangioleiomyomatosis often presents with dyspnoea and airflow obstruction, the diagnosis is often missed initially. The mean interval between onset of symptoms and diagnosis was over four years in our series (range 0–25 years) and the picture is similar elsewhere.2 The diagnosis should be suspected in women when dyspnoea or asthma is associated with a pneumothorax, haemoptysis or abnormal chest radiograph, when emphysema has been diagnosed without a history of smoking or α1-antitrypsin deficiency, or when interstitial lung disease occurs with airflow obstruction.
Open lung biopsy has been the gold standard for the diagnosis of lymphangioleiomyomatosis. The sensitivity and specificity of histological analysis has improved considerably with the use of the monoclonal antibody HMB 45 which, in the lung, will only stain LAM cells19 ,20 thus excluding conditions which may mimic lymphangioleiomyomatosis such as metastatic endometrial sarcoma.33 Open lung biopsy can determine the hormone receptor status of LAM cells and whether smooth muscle proliferation or cystic change predominates but, as the clinical significance of these findings is uncertain, they do not alone justify taking open lung biopsy specimens. Transbronchial biopsy specimens in conjunction with HMB 45 staining can be sufficient to make a diagnosis of lymphangioleiomyomatosis.20
The plain radiograph may be normal initially but, as the disease progresses, reticular shadowing and cystic changes develop whilst lung volumes are either preserved or increased (fig 3).31 ,34The association of large lungs and diffuse shadowing on the plain radiograph may also be seen in Langerhans’ cell histiocytosis, sarcoidosis, and extrinsic allergic alveolitis. The appearances on high resolution computerised tomographic (HRCT) scanning are characteristic and striking with thin walled cysts 2–20 mm or more in diameter distributed throughout the lung fields (fig 3).31 ,34Other features include occasional hilar and mediastinal nodes and alveolar shadowing, which may represent haemorrhage, in up to 60% of scans.6 ,25 ,34 The main radiological differential diagnosis is Langerhans’ cell histiocytosis which, unlike lymphangioleiomyomatosis, usually has a nodular appearance at least in the early stages and it spares the costophrenic angles.35


(A) Chest radiograph showing a reticular pattern which appears most prominent at the bases. (B) High resolution CT scan showing the classical findings of multiple thin walled cysts evenly distributed throughout the lung fields; the intervening parenchyma appears normal.
Because the CT findings in lymphangioleiomyomatosis are so distinctive, many physicians do not necessarily resort to open lung biopsy to make the diagnosis. This strategy is probably reasonable if the CT appearance is classical and the history is fully consistent with lymphangioleiomyomatosis; the finding of renal angiomyolipomas or a chylous effusion obviously strengthens the diagnosis. In practice, lung tissue often becomes available when patients undergo surgery for pneumothorax or chylous effusion.
MANAGEMENT
Supportive treatment
Patients with lymphangioleiomyomatosis and airflow obstruction are often helped by inhaled β agonists and a trial of treatment is warranted. Half the patients in our study were taking a β agonist and over half of those tested had more than 10% reversibility.36 Pneumothorax should be managed conventionally but, as it is more likely to be recurrent, bilateral, and less responsive to conservative measures, surgeons should be involved at an early stage. Recurrent pneumothorax will require pleural abrasion, talc or chemical pleurodesis, or pleurectomy. The possibility that patients may be considered for lung transplantation in the future needs to be borne in mind as all three procedures increase the risk of bleeding at the time of transplantation. Most patients now receive single lung transplants and, although full pleurectomy should be avoided if possible, neither pleurodesis nor limited pleurectomy precludes single or double lung transplantation. It may be advisable to consult a transplantation team prior to undertaking surgical pleural procedures, although decisions on treatment for the pneumothorax must be based primarily on the clinical needs of the patient at the time.
Chylous pleural effusions resulting from obstruction of the thoracic duct may require drainage, although this results in loss of protein and lymphocytes. Lymph formation can be reduced by dietary modification, either substituting dietary fat with medium chain triglycerides (which are carried in venous blood rather than lymphatics37) or a low fat diet. In practice a medium chain triglyceride diet is fairly unpalatable and a low fat diet may be more acceptable. Anecdotal reports suggest that patients with chylous collections are more likely to benefit from progesterone treatment.2 Pleurodesis and pleurectomy have been effective in troublesome cases of recurrent effusions and have been used satisfactorily in combination with thoracic duct ligation in two patients.
Patients often feel isolated when given the diagnosis of a rare disease and patient groups can be a helpful source of information and support, particularly when newly diagnosed. The LAM Trust (UK) and the LAM Foundation (USA) have been set up by and for patients with lymphangioleiomyomatosis and contact numbers are given at the end of this article. Both provide fact sheets for patients which may also be of use to doctors looking after patients.
Hormone therapy
Lymphangioleiomyomatosis is thought to be hormonally dependent and hormonal manipulation in the form of progesterone supplementation or anti-oestrogen measures has been widely used. There have been no controlled trials of treatment, however, and approaches vary considerably between countries. Data on the response to therapy come from case reports, a meta-analysis of 12 patients,38 and a retrospective assessment from three large series covering 128 patients, although only a small proportion had data suitable for analysis and the outcome measures were different in each study.2 ,4 ,6 As case reports are likely to over-represent treatment successes, more weight has been given to data from the larger series.
Progesterone: Progesterone is the treatment used most commonly in the UK and is usually given as intramuscular depot medroxyprogesterone 400–800 mg/month or, less frequently, as oral progesterone, 10–20 mg/day. The pharmacokinetics of the two preparations are quite different with plasma progesterone levels showing large fluctuations after each oral dose. Following intramuscular treatment plasma levels are threefold higher than with oral treatment, show little fluctuation, and continue to rise smoothly over the first few months but then plateau.39 ,40
The evidence that progesterone is effective in lymphangioleiomyomatosis is probably better than for other forms of treatment but it is still weak. In the three large retrospective studies performed to date2 ,4 ,6 32 patients had taken a mean daily dose of at least 10 mg progesterone orally or intramuscularly and in 15 patients the disease was thought to have improved or stabilised. In our retrospective review the mean decline in FEV1 appeared to be less in patients taking progesterone than in those receiving no treatment, although we cannot exclude the possibility that the findings were due to differences in baseline lung function or selection bias. In the meta-analysis of case reports progesterone treatment was considered successful in four of the eight patients.
Oophorectomy: Surgical oophorectomy is used to treat lymphangioleiomyomatosis38 ,41 although it has often been combined with progesterone42 ,43 and/or tamoxifen.44 Although it appeared to be the most successful intervention in the meta-analysis with some improvement in five of seven evaluable patients,38 only six patients in two of the larger studies2 ,6 had an oophorectomy alone of whom none improved although three appeared to stabilise. Chemical oophorectomy with gonadotrophin releasing hormone agonists45 has been used alone46 and in conjunction with other treatments but there are few data to assess its value.
Tamoxifen: The evidence for using tamoxifen as a single agent in lymphangioleiomyomatosis is weak. It has been found to stabilise the disease in five of 13 cases2 ,6 but to exacerbate it in others.47 Tamoxifen is a partial agonist at the oestrogen receptor and the newer full oestrogen receptor antagonists or aromatase inhibitors which block oestrogen synthesis may be more effective.
Occasional case reports have suggested benefit in response to other treatments including interferon-α48 and somatostatin49 but these are difficult to assess.
Trying to assess the value of treatment from retrospective surveys is unsatisfactory and prone to bias. Patients show transient fluctuation in symptoms and lung function when taking no treatment, making assessment of anecdotal reports difficult. Overall the evidence suggests that progesterone is the drug most likely to be of benefit although many patients do not respond. When it should be started and whether it should be given intramuscularly or orally is also uncertain. It is difficult to recommend tamoxifen on the available evidence and too few patients have had surgical oophorectomy alone to justify recommending this treatment. Gonadotrophin releasing hormone agonists are a possible alternative to surgical oophorectomy although there are few data on their use. Although the rarity of lymphangioleiomyomatosis makes new treatments difficult to evaluate, a randomised placebo controlled trial of progesterone in women with well preserved lung function would be possible with international collaboration and would seem to be the best way to make progress.
Lung transplantation
Lung transplantation, mainly of single lungs, has been carried out successfully in patients with end stage lymphangioleiomyomatosis and in a review of 59 cases worldwide the actuarial survival at two years (58%) was similar to the figures following transplantation for other lung diseases.25 The main problems relating to lymphangioleiomyomatosis or previous pleurectomy were intraoperative haemorrhage, resulting in one death and two repeat thoracotomies, and postoperative chylothorax and spontaneous pneumothorax in the native lung.25 Recurrent lymphangioleiomyomatosis has occurred in three cases in the donor lung (all male)50-52 and in one case this was confirmed with HMB 45 (a useful technique to differentiate recurrent disease from obliterative bronchiolitis).
Pregnancy
Patients with lymphangioleiomyomatosis are young and many wish to become pregnant. Although some patients have had uncomplicated pregnancies, several case reports suggest that pregnancy can exacerbate lymphangioleiomyomatosis.10-12 Our findings support this since, amongst the seven pregnancies that occurred when or after the diagnosis of lymphangioleiomyomatosis was made, five were complicated by a pneumothorax or chylous effusion requiring intercostal drainage and three required surgery.53 Patients should be warned that pregnancy may be associated with a higher incidence of respiratory complications and deterioration in their disease.53
Renal angiomyolipoma
Although renal angiomyolipomas are present in about 50% of patients with lymphangioleiomyomatosis,28 ,29 the majority are small and asymptomatic.54-56 Those greater than 4 cm in diameter and multiple tumours are more likely to grow more rapidly and cause bleeding, which usually presents as haematuria or flank pain.54 ,56 ,57 The CT appearance of angiomyolipomas is fairly characteristic, making biopsy specimens unnecessary in most cases.58 A warning, however, since a renal carcinoma can mimic the CT appearance of an angiomyolipoma59 and two patients with lymphangioleiomyomatosis have developed renal cancer.60 ,61 The findings on renal ultrasound are less specific for angiomyolipomas but can be used to follow progress.
There are no definitive guidelines on the management of angiomyolipomas but small asymptomatic tumours do not require treatment and can be followed by occasional ultrasound or CT scanning. Bleeding, pain and rapid growth are indications for treatment54 and some would advocate prophylactic treatment of asymptomatic larger tumours.55 Nephrectomy is sometimes performed when a malignant tumour is suspected but treatment should normally aim to conserve renal tissue as far as possible. Super-selective embolisation is becoming the treatment of choice55 ,56 since it conserves renal tissue, reduces rebleeding rates,62 and does not require general anaesthesia. Whether progesterone inhibits growth of angiomyolipomas is unknown.
Screening for tuberous sclerosis
Pulmonary lymphangioleiomyomatosis can occur as part of the tuberous sclerosis complex (TSC) and patients with pulmonary manifestations of TSC may not have the classical features such as epilepsy and mental retardation.63 TSC is an autosomal dominant condition associated with two genes, TSC-1 on chromosome 964 and TSC-2 on chromosome 16.65 Although the expressivity of TSC is variable, the disease has a high level of penetrance and making the diagnosis has profound implications for the patients and their families. The likelihood of a patient with lymphangioleiomyomatosis having TSC is greater in patients with an angiomyolipoma. The diagnosis of tuberous sclerosis is generally made clinically and criteria have been described by Gomez66 and the National Tuberous Sclerosis Association (NTSA).67 The presence of one primary feature, two secondary features, or one secondary and two tertiary features are required for a definite diagnosis of TSC. NTSA criteria are shown in table 1; angiomyolipomas and lymphangioleiomyomatosis are both secondary features and hence such patients would be labelled as having TSC by these criteria. Since none of the women with isolated lymphangioleiomyomatosis with or without renal angiomyolipoma (but with no other features of TSC) has had a child with TSC, they presumably do not a have germ line mutation in either TSC gene and in our view they should not be considered as having TSC. All women with lymphangioleiomyomatosis should, however, undergo a careful family history and clinical examination for stigmata of TSC and, in cases of doubt, should be seen by a clinical geneticist.
Diagnostic criteria for tuberous sclerosis complex67
Aetiology, current research and future directions
An increased awareness of lymphangioleiomyomatosis and the concerted efforts of patients is stimulating both clinical and basic research into the disease. The National Institutes of Health is funding a five year clinical programme at the National Heart, Lung and Blood Institute (Bethesda, Maryland, USA) which will follow women with lymphangioleiomyomatosis and is likely to provide important information on the natural history and management of the disease. Various groups are carrying out cellular and genetic research although much of this work is still in its early stages and has only been published in abstract form. The current areas of interest are summarised here and, where results are still provisional, future areas of research. are suggested
RELATIONSHIP BETWEEN LYMPHANGIOLEIOMYOMATOSIS AND THE TUBEROUS SCLEROSIS COMPLEX (TSC)
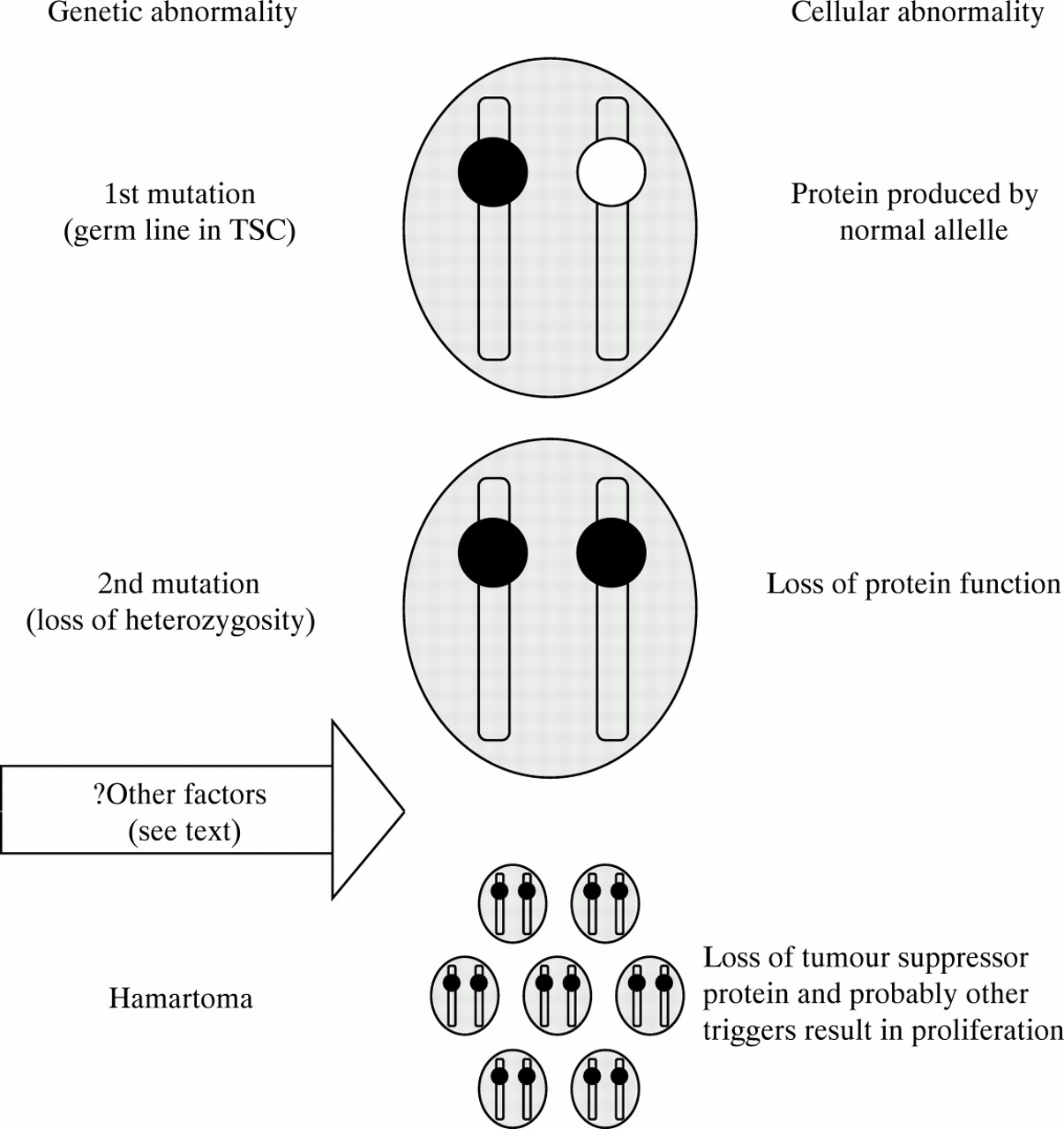
Lymphangioleiomyomatosis and angiomyolipomas can occur as part of TSC and understanding the differences between lymphangioleiomyomatosis and TSC may help to elucidate the aetiology of lymphangioleiomyomatosis. Although the link between the two diseases has been recognised for some years,1 ,28 ,29 ,63 the idea that TSC genes may be involved in lymphangioleiomyomatosis has only been explored more recently.68 ,69 In TSC there is a germ line mutation in one of two genes (TSC-1 or TSC-2).70-72 A second somatic mutation (or hit) termed loss of heterozygosity results in loss of the gene product in that cell (fig 4).73 As the TSC genes are tumour suppressors74 this allows cellular proliferation and the formation of a hamartoma; in TSC multiple second hits (mutations in the normal allele) result in multiple hamartomas.
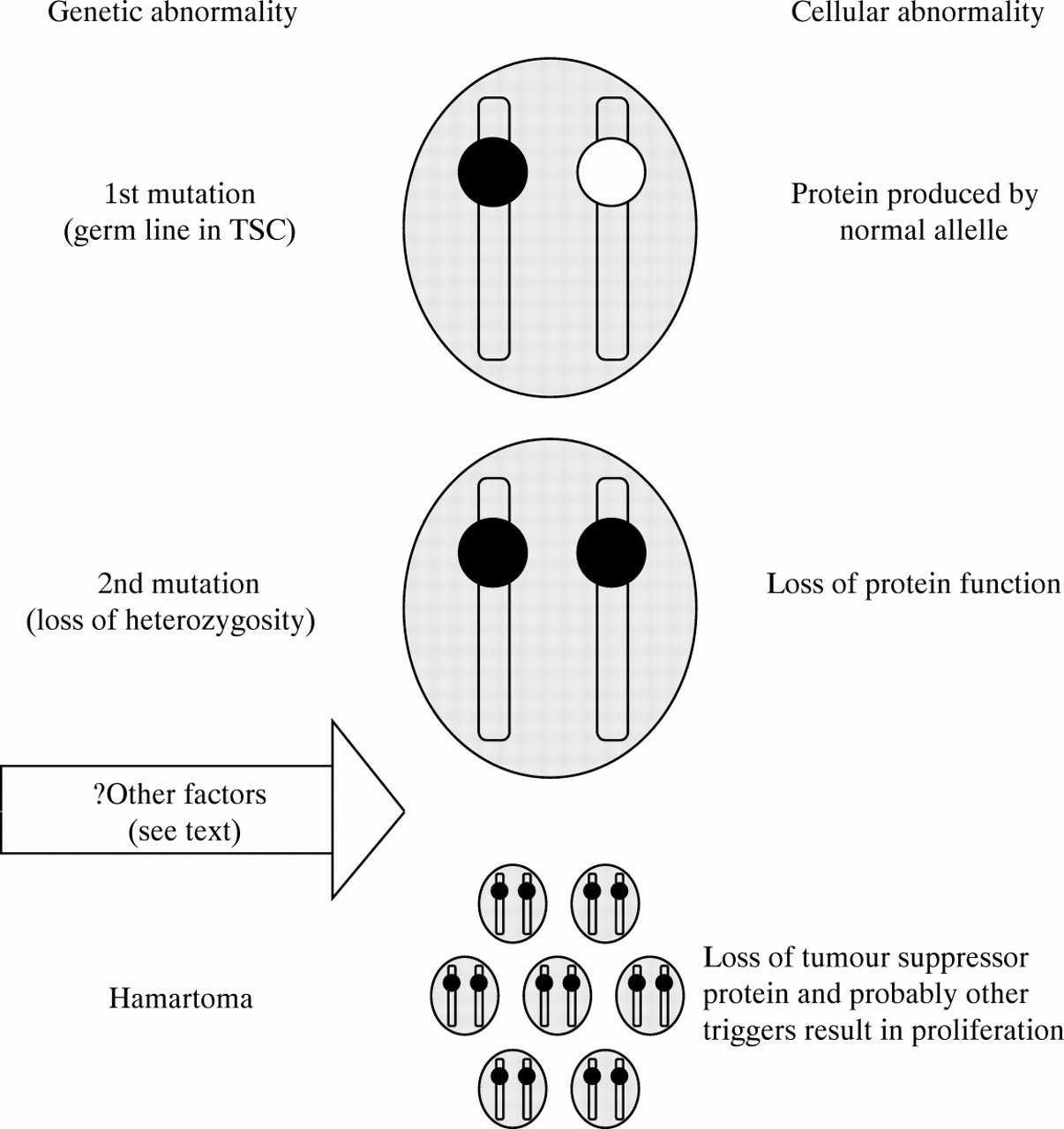
Schematic representation of how loss of heterozygosity for TCS-2 might occur in lymphangioleiomyomatosis. When the cell has one mutant allelle (black) normal protein is synthesised by the normal allelle (white). A second sequential somatic mutation results in loss of heterozygosity for that allelle and loss of protein function. Since TSC-2 is a tumour suppressor gene cellular proliferation results. In tuberous sclerosis the initial mutation is in the germ line: in lymphangioleiomyomatosis the initial mutation is unlikely to be in the germ line and probably represents somatic mosaicism.
An important advance in the genetics of lymphangioleiomyomatosis was made this year by Smolarek et al.69 They found loss of heterozygosity for TSC-2 (but not TSC-1) in renal angiomyolipomas from seven of 13 patients with lymphangioleiomyomatosis but with no cutaneous or neurological features of TSC, and in the lymph nodes of a further patient. Lung tissue was not examined, probably because the diffuse nature of the lung disease makes it difficult to separate the DNA from LAM cells and normal lung tissue to enable loss of heterozygosity to be identified. Loss of heterozygosity for TSC-2 had been shown previously in sporadic angiomyolipomas (without lymphangioleiomyomatosis or tuberous sclerosis),75 but the relatively high incidence of TSC-2 abnormalities in the study by Smolarek et al 69 and the fact that they also occurred in lymph nodes suggest that the findings are relevant to lymphangioleiomyomatosis. Since a germ line mutation of TSC-2 is unlikely in lymphangioleiomyomatosis, these patients may be mosaics—that is, they may only carry the TSC-2 mutation in a small population of cells or may have sequential somatic mutations of the TSC-2 gene.
CELLULAR ROLE OF THE TUBEROUS SCLEROSIS-2 GENE PRODUCT TUBERIN AND RELATED PROTEINS
The TSC-2 gene product tuberin is constitutively expressed in a large number of tissue and cell types and is therefore likely to perform a function of basic cellular importance. Tuberin is a 180 kDa protein which acts as a tumour suppressor73; it has guanine triphosphate (GTP) activating protein (GAP) activity for the GTP binding proteins rap 176 and rab 5.77 GTP binding proteins regulate cellular pathways and are active or inactive according to whether GDP or GTP is bound. GAPs such as rap 1 GAP and tuberin facilitate the hydrolysis (inactivation) of GTP attached to the GTP binding protein (fig 5A). Recent studies are beginning to shed light on the cellular role of tuberin in inhibiting rap 1, a modulator of the ras pathway, an important pathway linking cell surface tyrosine kinase receptors and the production of transcription factors involved in cell proliferation (fig 5B). Activation of tyrosine kinase receptors (by growth factors) results in activation of ras by the phosphorylation of intermediate proteins. This induces activation of raf-1 and thence a protein kinase cascade via mitogen activated protein kinases resulting in the upregulation of transcription factors and cellular proliferation. Rap 1 binds many of the same regulators and effectors as ras78 and, according to cell type, can either inhibit or stimulate proliferation: it reverses the transformation of NIH3T3 cells by the oncogene vKi ras79 but induces DNA synthesis when injected into Swiss 3T3 cells.80 Loss of tuberin may therefore cause cellular proliferation through one or more of the following mechanisms: (1) in cells where rap 1 promotes mitogenesis loss of tuberin would lead to activation of rap and thence proliferation; (2) in cells where rap 1 antagonises ras (is anti-mitogenic), loss of tuberin binding to rap may allow rap to associate with its other effectors to promote mitogenesis; (3) tuberin may have functions other than its rap GAP activity including GAP activity for other proteins or as an effector for non-GAP functions in these or other pathways.

(A) Regulation of guanylyl triphosphate (GTP) binding proteins. GTP binding proteins act as molecular switches being active when bound to GTP and inactive when bound to guanylyl diphosphate (GDP). GTP hydrolysis to the inactive form is promoted by specific GTP activating proteins (GAPs) including rap 1 GAP and tuberin. The active form is promoted by antagonistic guanine dissociation factors (GDF). GAPs themselves may also be GTP binding proteins. (B) Simplified representation of how the tyrosine kinase receptor (TKR)/ras/raf/protein kinase cascade leads to the production of transcription factors that are important in cellular proliferation. The figure shows the possible interactions of tuberin and rap 1 with unproven associations shown shaded. Binding of the TKR ligand activates protein phosphorylation via intermediate proteins to activate ras and thence raf 1. Further phosphorylation events occur downstream via mitogen activated protein kinase kinase (MAPKK) and mitogen activated protein kinase (MAPK) resulting in the upregulation of nuclear transcription factors and proliferation. Tuberin inhibits rap 1 which in some cells inhibits the ras pathway whereas in others it promotes mitogenesis. — denotes inhibition.
Evidence linking defects in the ras pathway to lymphangioleiomyomatosis is at present circumstantial. Depletion of tuberin protein by antisense oligonucleotides allowed serum depleted fibroblasts to enter the S phase of the cell cycle unlike tuberin replete cells. The response was similar to cells maximally stimulated by serum,81suggesting that cells depleted of tuberin may proliferate in the absence of mitogenic stimuli. In the Eker rat, an animal model with a germ line truncating mutation of TSC-2 used to study tuberous sclerosis, homozygous mutants die in utero whilst heterozygotes develop renal and uterine tumours. If wild type TSC-2 is introduced into these tumour cells their ability to proliferate is reduced.82 It has been argued that the GAP activity of tuberin for rap 1 is weak,76 but there seems little doubt that the tumour suppressor activity of tuberin is due to its rap 1 GAP activity since the region homologous with rap 1 GAP showed similar tumour suppressor activity to the full length protein.82 There are parallels between this hypothesis and neurofibromatosis (NF) where the NF 1 gene product neurofibromin has GAP activity for ras.83 NF tumour cells express very little neurofibromin and their ras proteins appear to be constitutively activated as judged by GTP binding.83
The other known function of tuberin is as a GAP for rab 5, one of the rab proteins that regulate intracellular transport. Rab 5 is the rate limiting step in the docking and fusion process of the endocytic pathway84 and there is evidence to suggest that tuberin may inactivate rab 5 in vivo, thereby reducing endocytosis. Immune complexes of both recombinant and native tuberin have rab 5 GAP activity. Rab 5 GAP activity is low in tuberin deficient cells and increases following the introduction of tuberin as does the rate of fluid phase endocytosis.77 Whether tuberin is a more important GAP for rap 1 or rab 5 in LAM cells and which of these two functions is more relevant to lymphangioleiomyomatosis is uncertain. We need to know more about the activities of rap 1 and rab 5 in LAM and normal smooth muscle cells and the relative GTPase activity of tuberin for these two proteins.
TSC-1 was sequenced in 1997.64 The predicted protein, named hamartin, also appears to have tumour suppressor activity since most of the genetic abnormalities described in hamartomas of TSC patients with TSC-1 mutations have been truncating mutations which are likely to cause loss of protein function.64 Hamartin co-localises with tuberin in cells and the two proteins interact via coiled coil domains85 so it is possible that a tuberin/hamartin complex is required for normal cellular function. This could explain the virtually identical phenotypes seen in patients with TSC irrespective of genotype. Loss of heterozygosity for TSC-1 is seen less frequently in TSC hamartomas and has not been described in lymphangioleiomyomatosis,69although it might be expected to give rise to lymphangioleiomyomatosis in the same way as loss of heterozygosity for tuberin.
ROLE OF OESTROGEN AND PROGESTERONE IN LYMPHANGIOLEIOMYOMATOSIS
Hormonal factors appear to play a part in both the initiation and progression of lymphangioleiomyomatosis as evidenced by the female predilection for the disease, the finding of hormone receptors on some LAM cells, and the suggestion that exogenous oestrogens9and pregnancy may exacerbate the disease whilst progesterone treatment may reduce its progression. Furthermore, although there is no sex difference in TSC, the 3% of patients with TSC who have lymphangioleiomyomatosis are predominantly, if not exclusively, women.3 ,86 The mean age of onset for TSC in these patients was 16 years whereas the onset of lung disease was 33 years, similar to that seen in isolated lymphangioleiomyomatosis.3 These differences suggest that factors in addition to an absence of tuberin are required for the development of lymphangioleiomyomatosis and these may be hormonal.
How sex hormones might affect the development and progression of lymphangioleiomyomatosis is speculative. Oestrogen may amplify the effect of a mutant protein by downregulating tuberin or possibly other proteins with GAP activity which would otherwise compensate for a defect in tuberin activity. Differential expression of such proteins would explain the predilection of these lesions for certain tissues. Some LAM cells express oestrogen and/or progesterone receptors which may be important in cell growth. LAM cells which express oestrogen receptors also express the protein Bcl-2 which acts as a suppressor of apoptosis.87 Increasing the ratio of anti-apototic proteins such as Bcl-2 to its pro-apototic homologue Bax would reduce apoptosis of LAM cells and may represent a way in which oestrogen potentiates lymphangioleiomyomatosis. The biological significance of oestrogen and progesterone receptors and why they are not uniformly expressed on LAM cells needs to be explained, however.
ROLE OF MELANOMA PROTEINS EXPRESSED IN LAM CELLS
The monoclonal antibody HMB 45 is used by pathologists to diagnose malignant melanoma; the discovery that it stains LAM cells has led to considerable interest in its target proteins and their role in LAM cells.88 HMB 45 binds to two 10 kDa glycoproteins (Pmel 17 and GP100) which are expressed by malignant melanoma and other cell lines of melanoma origin. HMB 45 positivity and other ultrastructural features suggest a developmental relationship between lymphangioleiomyomatosis, angiomyolipoma, and the rare clear cell lung tumour (which can also be associated with TSC and lymphangioleiomyomatosis).18 ,89-92 This relationship is not shared by other hamartomas in TSC, however,93 which suggests that they have a different aetiology or that lymphangioleiomyomatosis and angiomyolipoma require an additional factor for their development. The Pmel 17 gene product controls the synthesis of eumelanin94 which is localised to pre-melanosomes in melanoma cells and structurally similar electron dense membrane bound vesicles are seen in LAM cells. HMB 45 negative LAM cells stain for proliferating cell nuclear antigen, a marker of proliferation, whereas HMB 45 positive cells do not,95which suggests that they may not be actively proliferating and that different populations of LAM cells have different functions. Whether expression of the HMB 45 ligand is important in the pathogenesis of lymphangioleiomyomatosis or a bystander phenomenon is uncertain.
ABERRANT EXPRESSION OF GROWTH AND TRANSCRIPTION FACTORS
Proliferation of normal cells may be enhanced by growth factors—whether autocrine, paracrine or circulating—and by increased or aberrant receptor expression. The diffuse and widespread nature of the smooth muscle proliferation seen in the lung in lymphangioleiomyomatosis and the recurrence of disease in the donor lungs of transplant recipients, including the apparent transformation of male donor cells,50-52 suggests that circulating factors may be important. Normal airway smooth muscle cells are capable of producing a large number of growth factors and other mitogens96 and a number of abnormalities in growth and transcription factors have been described in LAM cells in the last two years (at present only in abstract form). These include: (1) increased expression of basic fibroblast growth factor97 and platelet derived growth factor β-chain and receptors98in biopsy specimens from patients with lymphangioleiomyomatosis; (2) increased production of angiotensin II in LAM cells99 and increased circulating levels of angiotensin converting enzyme in patients. This is of interest since angiotensin II upregulates transcription factors that cause hypertrophy of normal airway smooth muscle cells.100
High mobility group protein I-C (HMGI-C) was expressed in lung biopsy specimens of four patients with lymphangioleiomyomatosis but not in control subjects.101 The HMG proteins are a family of chromatin associated proteins that regulate gene expression in undifferentiated embryonic cells102 ,103 and they are frequently aberrant or overexpressed in uterine leiomyomas104 and pulmonary hamartomas.105Overexpression of aberrant HMGI-C transcripts in LAM cells may permit uncontrolled cellular proliferation.
Some of these phenomena could be secondary to uncontrolled cellular proliferation resulting from a defect in TSC-2 but where does the TSC-2 hypothesis fit in with the recurrent and widespread smooth muscle proliferation seen in donor lungs after transplantation? One possibility is that clonal expansion of one cell type secreting mitogenic factors leads to recruitment of a second reactive cell population.
Future directions
The most pressing clinical issue in the management of lymphangioleiomyomatosis is to determine the place of hormonal therapy. We need to establish whether progesterone does slow the course of the disease and, if so, should it be instituted at diagnosis? Should it be given orally or intramuscularly and what is the role of other treatment? These questions should be addressed by placebo controlled randomised trials in patients with well preserved lung function. Power calculations suggest that a crossover trial would require 50 to 100 patients to have reasonable power to detect a 50 ml difference in change in FEV1 over one year.4 International collaboration would therefore be required but this should now be possible.
At a cellular level confirmation of loss of heterozygosity of TSC-2 in lung tissue is required. Understanding the biology of tuberin in normal smooth muscle and LAM cells and its interaction with hormones and growth factors is clearly important. The products of LAM cells and their effect on neighbouring pulmonary cells may also be a fruitful area of research. Research would be helped considerably if current efforts to produce a pure LAM cell line were successful.
Although there is still much to learn about the clinical management and molecular biology of this rare and fascinating disease, we can expect genuine advances in understanding the aetiology and, hopefully, the management of lymphangioleiomyomatosis in the next few years. Because the biological research is focusing on basic cell function, we may also obtain insight into mechanisms that are of relevance to other diseases that involve smooth muscle hyperplasia.
Acknowledgments
I would like to thank Professor Anne Tattersfield for her helpful comments on the manuscript, Dr Paul Corris for advice on transplantation, Dr Colin Clelland for the pathology slides, Mr W E Morgan for the operative photographs, and the LAM Trust (UK), the LAM Foundation (USA), and the British Lung Foundation for financial support.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}