Article Text

Statistics from Altmetric.com
Primary pulmonary hypertension (PPH) is a rare disorder with an incidence of one to two cases per million people per year in Western populations. It is a progressive disease usually affecting the arterial side of the pulmonary circulation and, if untreated, progresses to severe pulmonary hypertension and, finally, right heart failure. Characteristically, and because of the vague nature of the symptoms, PPH may take up to 18 months to diagnose and, following diagnosis, the median survival is under two years. The disease is most common in women (ratio 1.7:1) and the mean age at the time of diagnosis is in the mid 30s. This bleak picture of PPH has been transformed over the last decade, partly because of better diagnostic techniques allowing earlier diagnosis and also because of new treatments, in particular the use of continuous intravenous prostacyclin. It is now realised that vasoconstriction of the pulmonary vascular bed is not the primary cause of PPH but occurs in association with marked histological changes. These histological changes are seen not only in PPH but also in severe pulmonary hypertension in association with other disorders such as HIV infection, connective tissue disease, and the use of appetite suppressant drugs. The realisation that primary or unexplained pulmonary hypertension was the end of a spectrum of several diseases causing severe pulmonary hypertension has lent a new impetus to the understanding of the disease and prompted a major symposium sponsored by the World Health Organisation held in Evian, France, in September 1998. At this symposium were gathered clinical scientists from all over the world with an interest in pulmonary hypertension and the result was a consensus about classification of pulmonary hypertension, the pathobiological methods of screening, risk assessment, and treatment. The first part of this review considers the clinical aspects of pulmonary hypertension, in particular the classification of pulmonary hypertension, the pathology, methods of screening for pulmonary hypertension, assessment of patients who are known to have pulmonary hypertension, and treatment of the disease. The second part focuses on what is and is not known about the pathobiology of pulmonary hypertension and how clinical and basic science may lead us to a further understanding of this enigmatic series of conditions. It is clear that the future is exciting in the world of pulmonary vascular medicine, and we are probably at the threshold of major new developments in pathobiological understanding and the treatment of vascular disease which may have ramifications outside the world of the lung.
Clinical
CLASSIFICATION OF PULMONARY HYPERTENSION
In the past, pulmonary hypertension has been classified as either primary (unexplained) or secondary. However, included in secondary pulmonary hypertension were a wide ranging number of causes such as left heart dysfunction, hypoxic lung disease, left to right shunt, and liver disease. This classification was therefore unsatisfactory, particularly when it was realised that both the pathological abnormalities and the treatment may be similar in patients with a severe pulmonary hypertension from whatever cause. This realisation has prompted a new nosology and a new classification. The new classification proposed at the WHO meeting in 19981 is shown in table 1.
WHO classification of pulmonary hypertension1
RISK FACTORS FOR PULMONARY HYPERTENSION
From the classification shown in table 1 it is clear that there are various risk factors for severe pulmonary hypertension. However, it is extremely important to assess the level of risk and hence the likelihood that a patient with a given condition will develop pulmonary hypertension. This is important both on the grounds of conservation of resources and cost. A detailed analysis of the pathobiology and suggested mechanisms of pulmonary hypertension is presented in the next section but the group at the WHO provided a useful classification of risk which can be used as a basis for screening patients who may develop pulmonary hypertension (table 2). We can define a risk factor for pulmonary hypertension as a factor that may either be necessary for causation or facilitate the development of the disease. Sometimes, of course, it is not possible to determine whether a factor was present prior to the onset of pulmonary hypertension, in which case it is described as an associated condition—that is, a condition that co-exists with pulmonary hypertension and therefore implies a common risk factor. The Risk Factor Subcommittee headed by Lucien Abenheim rated risks on the basis of the strength of their association with pulmonary hypertension. Definite indicates an association based on several different observations and often including a major study; very likelyindicates a similar series of observation or a general consensus amongst experts; possible indicates an association based on case reports, registries such as the National Institute of Health Registry of Primary Pulmonary Hypertension, and expert opinions; and unlikely indicates risk factors that have been proposed but for which no additional evidence has been forthcoming.
WHO classification of risk for pulmonary hypertension1
SCREENING FOR PULMONARY HYPERTENSION
Since PPH usually takes two years to diagnose, median survival is only a further two years, and the prognosis probably depends on making a diagnosis when the pulmonary circulation is compliant and responsive to vasodilator therapy, it is very important to try to make the diagnosis as early as possible. On the other hand, a full assessment of the pulmonary circulation involves right heart catheterisation which is an invasive procedure. Clearly, a balance has to be drawn between the need to make an early diagnosis (the benefits of early use of vasodilators, though likely, are not yet established) and the risks of the diagnostic techniques that are to be used. The advent of widely available echocardiography has transformed our ability to screen patients for pulmonary hypertension. Other techniques such as cardiopulmonary exercise testing, magnetic resonance imaging, and spiral CT scanning are also likely to play an increasing part in the future in the diagnosis of pulmonary hypertension.
Who to screen
If we are to screen for pulmonary hypertension it is very important to focus screening on those who are most likely to have the disease and most likely to benefit from treatment of the disease. Major associations with pulmonary hypertension are as follows.
(1) Family history. The genetics of pulmonary hypertension is dealt with below but it has been clear for some time, since the early studies in Nashville, USA, that a significant proportion of patients (approximately 6–10% of cases) have a family history of pulmonary hypertension. There are probably at least two genes of which the first to be discovered was PPH 1 (locus 2q 31–32). Familial pulmonary hypertension is inherited as an autosomal dominant with incomplete penetrance, suggesting that screening at least first degree relatives may be fruitful.2 It is suggested that first degree relatives should be screened by transthoracic echocardiography at the time of the diagnosis of the index case and at any time in the future should they develop symptoms consistent with pulmonary hypertension.
(2) Connective tissue disease. Some 10% of patients with PPH have Raynaud's phenomenon. It is also known that patients with connective tissue disease—in particular the CREST variant of systemic sclerosis but also other conditions such as rheumatoid arthritis and systemic lupus erythematosus (SLE)—are susceptible to pulmonary hypertension and, indeed, up to 40% of patients with CREST will die of the pulmonary hypertension.3 It is therefore recommended that transthoracic echocardiography is performed annually in patients with systemic sclerosis and those with other connective tissue diseases are screened should they develop appropriate symptoms.
(3) Appetite suppressants. It is ironic that the second WHO symposium on PPH should be held at the time of the second epidemic of pulmonary hypertension associated with appetite suppressant usage. The first WHO symposium 25 years ago was held following an epidemic of pulmonary hypertension in Europe in association with the appetite suppressant agent aminorex fumarate.4 The more recent association between appetite suppressant drugs and pulmonary hypertension has occurred since the widespread use of fenfluramine and dexfenfluramine. Perhaps, luckily, the association was discovered quite early and the prevalence is not nearly as high as it was for aminorex.5At present we do not believe it is useful to screen patients who have taken an appetite suppressant for resting pulmonary hypertension since the incidence is low (approximately 1 in 20 000 among a population who have used the drugs for three months) even though the relative risk is high.
(4) Portal hypertension. Patients with liver disease may develop either right to left shunting6 or pulmonary hypertension. Pulmonary hypertension seems only to occur where there is also portal hypertension, suggesting that cirrhosis on its own is not sufficient.7-9 It is only necessary to screen patients with cirrhosis if they are being considered for liver transplantation, in which case it becomes mandatory.
(5) HIV infection. The association between pulmonary hypertension and HIV infection has been known for a number of years.10 ,11Since these patients will probably have several other important reasons for ill health, it is not recommended that they are screened for pulmonary hypertension but they should be offered echocardiography if appropriate symptoms develop.
(6) Drug use. There seems to be an association between bothl-tryptophan and 5-hydroxytryptamine uptake inhibitors and pulmonary hypertension. The mechanisms are not understood and at present no recommendation can be made about screening.
(7) Thyroid disease. There is a known link between underactive thyroid and PPH.12 It is not recommended that patients with hypothyroidism are screened for pulmonary hypertension but clearly patients with pulmonary hypertension should have thyroid function tests.
How to screen
Clearly, it is not appropriate for all patients at risk to have a transthoracic echocardiogram. In low risk individuals particular attention should be paid to clinical features that might indicate the development of pulmonary hypertension, though these are often enigmatic, particularly when patients have other more urgent symptoms and signs associated with their primary disorder. Typically, patients complain of breathlessness on exertion with no other important symptoms and few cardiopulmonary signs. The clinical symptoms and signs of pulmonary hypertension are described below but, unfortunately, by the time the typical clinical features develop, there is usually advanced disease so a patient with exertional breathlessness of no known cause and few clinical signs should be considered as having pulmonary hypertension until proved otherwise. Basic additional investigations should include the following:
(1) Chest radiograph: this may show an enlarged heart and enlarged main pulmonary artery. The experienced observer may notice peripheral pruning of the pulmonary vasculature.
(2) Electrocardiogram: this may show evidence of right atrial and right ventricular hypertrophy, right axis deviation and strain.
(3) Blood tests: these depend on the history; there may be a need to measure HIV status, thyroid function, autoantibodies, or to look for evidence of underlying liver disease.
(4) Transthoracic echocardiogram: this non-invasive investigation is the screening method of choice and is widely available in most centres. It will demonstrate the presence of pulmonary hypertension (by measure of tricuspid regurgitant jet velocity and relationship of acceleration time to ejection time in the pulmonary outflow tract), right ventricular overload (increase in right ventricular size), and other indices (see below). It may also help to determine the underlying diagnosis by showing the presence of clot, shunt, or evidence of left ventricular disease. A weakness of echocardiography is that the investigation is usually done at rest whereas at least 70% of pulmonary circulation must be obstructed before the resting pulmonary artery pressure rises. It is likely in the future that echocardiography will be enhanced and amplified by making measurements on exercise or following passive leg raising which increases venous return to the right heart.13 If pulmonary hypertension or evidence of right ventricular dysfunction is discovered on echocardiography, the next consideration is how often to repeat the test or, indeed, whether to proceed to right heart catheterisation. These questions have not been answered but in high risk individuals it would seem reasonable to repeat the echocardiogram every year and to arrange cardiac catheterisation when the pulmonary artery systolic pressure as measured by echocardiography rises above 40 mm Hg.
PATHOLOGY OF PULMONARY HYPERTENSION
The pulmonary vasculature has both similarities to and differences from the systemic circulation. The similarities include an arterial structure which consists of an intima composed of endothelial cells, a media composed of smooth muscle cells, and adventitia composed of protein matrix secreted by adventitial fibroblasts. A major difference between the two circulations is that the pulmonary circulation is a high flow, low pressure system with a remarkable degree of compliance to cope with cardiac outputs that may rise sixfold with heavy exercise. Furthermore, the pulmonary vessels must be thin walled to allow gas exchange but must be sufficiently well supported so as not to rupture. A further important difference between the two circulations is the response to hypoxia. The systemic circulation dilates to hypoxia whereas the pulmonary circulation constricts. Hypoxic pulmonary vasoconstriction is a reflex that has been known about since 194614 but is still not fully understood, nor is the relationship between the vasoconstriction/remodelling that occurs following hypoxia and that which accompanies PPH. At present the consensus is that hypoxic pulmonary vascular damage is different. However, patients with severe pulmonary hypertension (which is rare in pulmonary hypertension caused by hypoxia) have underlying histological abnormalities which may vary slightly depending on aetiology but show a remarkable degree of similarity.
PPH is characterised by intimal fibrosis, in situ thrombosis, and hypertrophy of the smooth muscle cells of the media and is divided into three pathological types: plexogenic arteriopathy, thrombotic arteriopathy, and pulmonary veno-occlusive disease.15 The plexogenic form (approximately 50% of cases) has the worst prognosis and is associated with the plexiform lesion, an unusual pathological entity consisting of a mass of disorganised vessels associated with endothelial cells, smooth muscle cells, and myofibroblasts. This may represent an angiogenic response to underlying vascular injury. In the thrombotic form there is similar pulmonary vascular histology but, in addition, multiple thrombi are present. These thrombi give the abnormal patchy appearance seen in a ventilation perfusion scan and may be a consequence of endothelial injury. They may also explain why anticoagulants improve survival in these patients. Pulmonary veno-occlusive disease (less than 10% of patients) appears to be a separate entity with intimal proliferation of the intrapulmonary veins rather than arteries. Although very rare, it is important to make the diagnosis because treatment with intravenous vasodilators such as prostacyclin can be fatal. The mechanisms underlying the pathological appearances seen in severe pulmonary hypertension are not fully understood but are considered below.
ASSESSMENT OF PULMONARY HYPERTENSION
Clinical assessment of pulmonary hypertension involves three separate intellectual processes. Firstly, we need to look for and understand the cause of the pulmonary hypertension; secondly, we need to evaluate pulmonary haemodynamics and their effect on pulmonary and cardiac function; and thirdly, we need to assess patients for responsiveness to vasodilator therapy whether by oral calcium channel blockers or by continuous intravenous vasodilators.
Making the diagnosis
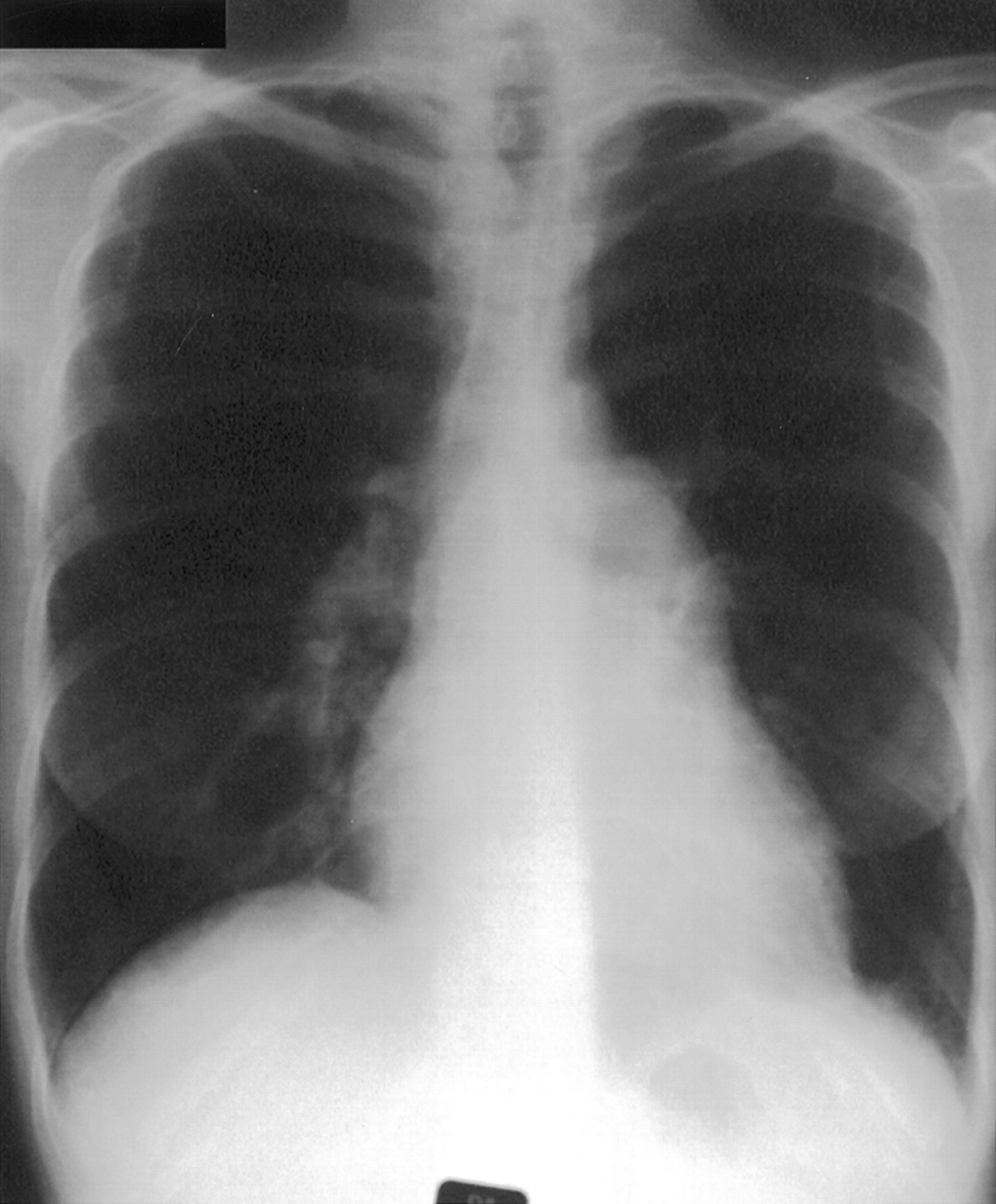
Important clinical features include breathlessness with no other obvious cause, specific signs of pulmonary hypertension (loud pulmonary second sound, right ventricular heave, pulmonary flow murmur), signs of right heart dysfunction (raised jugular venous pressure, V waves in the neck, hepatic pulsation, swollen ankles) and signs of co-existent disease (primary pulmonary, cardiac, or liver disease or evidence of venous thrombosis or connective tissue, thyroid or HIV disease). Chest radiography (fig 1) is of limited benefit but may show enlargement of the heart or main pulmonary artery or peripheral pruning of vessels. Electrocardiography may show right atrial or right ventricular hypertrophy, right axis deviation, and strain. Pulmonary function tests may indicate underlying lung disease but in “pure” pulmonary hypertension spirometric tests and lung volumes are usually normal but the carbon monoxide transfer factor is usually low, consistent with a low pulmonary capillary blood volume. The ventilation-perfusion scan (fig 2) may show the presence of mismatched defects indicating possible underlying thromboembolism, but defects may also be seen in pure intrinsic vascular disease, particularly the thrombotic variant. The ventilation scan will be normal but the perfusion scan may show patchy loss consistent with vascular obstruction. Computed tomographic (CT) scanning is needed to exclude parenchymal lung disease but volume scans of the central pulmonary arteries after injection of contrast may help to demonstrate major intravascular clot (fig 3). Echocardiography is the most useful non-invasive investigation. Transthoracic echocardiography will show a dilated right heart or right ventricular hypertrophy with evidence of pulmonary hypertension such as a decrease in the ratio of acceleration time to ejection time or an increase in tricuspid regurgitant wave velocity. In addition, echocardiography will show any left to right shunts and in the future it may be used to evaluate right ventricular performance by an index of myocardial performance.16-19
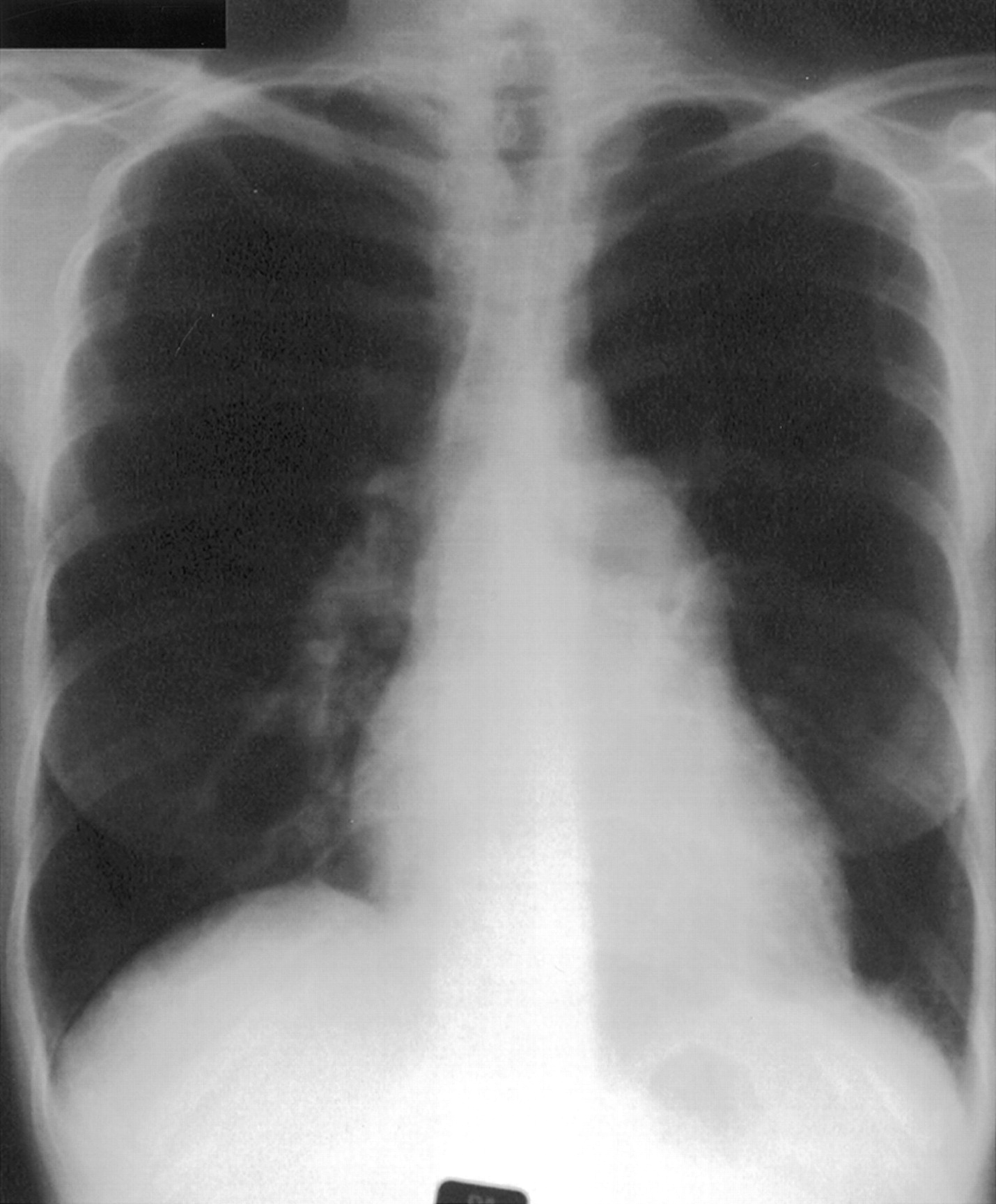
Chest radiograph from a patient with PPH showing the enlarged main pulmonary artery and enlarged heart.

Ventilation-perfusion scan from the patient in fig 1showing (A) normal ventilation but (B) patchy loss of perfusion due to vascular obstruction. Pulmonary embolism should be excluded.

Computed tomographic (CT) angiogram of the patient in fig 1 showing large pulmonary arteries and absence of intravascular clot.
Echocardiography is measured at rest and it would be useful to make echo-doppler measurements on exercise but this remains difficult. Cardiopulmonary exercise testing has been examined. The simplest cardiopulmonary exercise test is a six minute walk and the results correlate with pulmonary haemodynamics and long term survival.20 It has also been claimed that measurements of gas exchange during exercise correlate with pulmonary artery pressure.21 ,22 Other non-invasive techniques which may be of use include magnetic resonance imaging (MRI) to evaluate right ventricular function.23
All these non-invasive investigations are useful in helping to point to the underlying diagnosis and the extent of pulmonary hypertension. However, for definitive assessment right heart catheterisation is necessary. This should only be done at a designated pulmonary hypertension centre (see below) since the investigation and, in particular, the administration of vasodilators carries a significant risk in inexperienced hands. It is usually possible to conduct all relevant investigations at a single sitting, reducing the necessity for further catheterisation. These should include exclusion of shunt, measurement of pressures and cardiac outputs, pulmonary angiography, and measurement of vasodilator responsiveness. Right heart catheterisation is usually performed using a Swan Ganz triple lumen thermodynamic catheter inserted under fluoroscopic control via the right femoral vein. These catheters may be difficult to manipulate through a hypertrophied right heart and are not easy to pass up the superior vena cava in order to measure oxygen saturations at different positions in the right heart to identify a shunt. A multipurpose catheter is usually used for this purpose. The following pressures are measured: right atrial pressure, right ventricular pressure, pulmonary artery pressure, and pulmonary artery occlusion pressure. These measurements should be made with the glottis open at functional residual capacity to reduce the effect of intrapulmonary pressure swings on pressure. Cardiac output is measured in triplicate by thermodilution and the measurements are repeated after administration of 100% oxygen if there is concomitant hypoxaemia and after exercise if pressures appear to be normal. Selective pulmonary angiography is also performed should there be any doubt from the ventilation-perfusion scan about the presence or absence of thromboembolism (fig 4).

(A) Right and (B) left selective pulmonary angiograms from the patient in fig 1 showing large central pulmonary vessels but peripheral pruning and no evidence of pulmonary embolism.
This procedure carries an increased risk in patients with pulmonary hypertension because of sudden rises in pressure in hypertensive non-compliant vessels and the flow rates of contrast medium are usually reduced when pulmonary hypertension is present. Finally, a vasodilator study is done with continuous measurement of systemic arterial pressure and pulmonary artery pressure, heart rate, and heart rhythm while the vasodilator is administered. Vasodilator studies are risky in inexperienced hands because, if pressures start to fall, patients can undergo cardiac arrest from which they often cannot be resuscitated. It is therefore important to be able to recognise the early signs of a dangerous change in haemodynamics such as fall in systemic pressure, rise in heart rate, or failure of cardiac output to rise. Several vasodilators have been used including inhaled nitric oxide,24 adenosine,25 ,26 high dose calcium antagonists27 and prostacyclin,28 with adenosine, nitric oxide, and prostacyclin being the most popular for repeat vasodilator trials.29 Nitric oxide and prostacyclin25 ,30 ,31 have the advantage of being very short acting so their effects can be reversed should the patient's condition deteriorate. Ideally, following a vasodilator there should be a fall in pulmonary artery pressure accompanied by a rise in cardiac output. This is not always the case and often there is a rise in cardiac output with no change in pulmonary artery pressure. The significance of these haemodynamic changes is not fully understood but, where there is an appropriate haemodynamic response—that is, a fall in pulmonary artery pressure with a rise in output—the prognosis is much better. Unfortunately, only 30% of patients fall into this category.
All the investigations of pulmonary hypertension are done with the patient resting. However, since the usual complaint is of exertional breathlessness, there may be mechanisms for assessing a patient's response to exercise and the effect of treatment on that response in the future. Promising methods include cardiopulmonary exercise testing (see above) and continuous pulmonary artery pressure measurement using the new manometer tipped catheters.32
TREATMENT OF PULMONARY HYPERTENSION: GENERAL POINTS
Treatment of pulmonary hypertension centres round the treatment of an underlying cause if present and treatment of the pulmonary hypertension itself. The underlying causes that should be recognised and treated include hypoxic lung disease, thromboembolic pulmonary hypertension, HIV infection, thyroid dysfunction, connective tissue disease, left to right shunts, and left ventricular dysfunction. If underlying causes have been searched for and excluded or treated, an assessment must be made of the risks versus benefits of treating the pulmonary hypertension itself. The current consensus would be that treatment of mild pulmonary hypertension (mean pressure less than 30 mm Hg) is probably not advisable with the current therapeutic agents. Where severe pulmonary hypertension exists, the choice of treatment lies between calcium channel blockers and continuous intravenous prostacyclin together with anticoagulants.
TREATMENT OF SEVERE PULMONARY HYPERTENSION
This section deals only with severe intrinsic pulmonary vascular disease—that is, where the disease affects the vessels rather than a vascular response to other disease; the treatment of chronic obstructive pulmonary disease, left heart dysfunction, or left to right shunt will not be discussed. It concentrates on the treatments that have been evaluated for PPH, pulmonary hypertension associated with connective tissue disease, and pulmonary hypertension in association with portal hypertension. By far the greatest literature is in PPH where the issues surrounding treatment have become somewhat clearer and a consensus is beginning to emerge.33 The theoretical basis for the use of vasodilators in pulmonary hypertension has been reviewed elsewhere34; this paper concentrates on studies that have shown benefit in the pulmonary circulation including anticoagulants, vasodilator therapy, immunosuppressive therapy, inotropes, and the surgical creation of intracardiac shunt for therapeutic purposes.
Anticoagulants
It has been known for a number of years that long term treatment with warfarin improves the prognosis in PPH. This is true even for patients who do not respond to conventional vasodilators. For example, Rich et al showed that anticoagulants doubled survival in non-responders from 31% to 62% at three years.35 When anticoagulants are combined with a vasodilator they provide an additional benefit so it is standard practice in pulmonary vascular units to give anticoagulants to patients with resting pulmonary hypertension (usually mean PAP >30 mm Hg) unless there is a contraindication. The benefits of treating very early pulmonary hypertension are not yet established (see above). The reason for the benefit of anticoagulants is not clear, although it is presumed that they prevent in situ thrombosis which occurs particularly in the thrombotic variant of PPH and is seen in lung tissue at necropsy in patients dying from pulmonary hypertension.36 This raises the issue of the difference between so-called “chronic thromboembolic pulmonary hypertension” and “primary pulmonary hypertension with thrombosis”, but that distinction is outside the scope of this review. The goal of treatment should be to achieve an international normalised ratio (INR) of 2.
Since we know that prostacyclin can increase exercise tolerance over that seen with anticoagulants without causing vasodilation (see later), it is possible that the improved exercise capacity is due to its anti-platelet activity in which case other forms of anti-thrombotic agents may be useful in the future. There is some evidence that thromboxane synthetase inhibition may be helpful37 and perhaps other agents will provide even greater benefit, in particular the newer heparins and heparin-like compounds.
Calcium channel blockers
Over the last 10 years calcium channel blockers have been shown to improve morbidity and survival in patients with PPH.27 ,38Calcium channel blockers are a heterogeneous group of compounds but it is the dihydropyridine blockers that have been used in patients with pulmonary hypertension, the rationale being that their principal action is on vascular smooth muscle. Their depressant effect on myocardial contractility is the principal limitation to their use. They are used in very high doses—for example, diltiazem up to 720 mg daily and nifedipine up to 300 mg daily. Although studies have shown that high doses are necessary, proper dose response relationships have never been shown and it is difficult to choose a dose for a given patient. The drugs are potentially dangerous and should not be given without formal vasodilator studies. Only patients who respond to acute vasodilator testing should be treated with a calcium antagonist. Once treatment is started, patients must be very closely monitored in hospital to ensure that they do not deteriorate. Patients may develop heart failure and, if this is not identified early, they can deteriorate rapidly. For these reasons treatment with a calcium antagonist should ideally only be initiated by those with appropriate experience in a pulmonary vascular unit.
The mechanism of action of calcium antagonists is presumed to be that of vasodilation but they may have other actions such as reduction in smooth muscle cell replication or effects on cell growth of other pulmonary vascular cells. Adverse effects of calcium antagonists include systemic hypotension, hypoxaemia due to increased ventilation-perfusion mismatching, right ventricular dysfunction, cardiogenic shock, arrhythmias, and death. However, there is now good evidence that calcium antagonists in combination with anticoagulants can produce five year survivals of the order of 90% in patients who respond to an acute vasodilator trial, so it appears that they may slow or abolish the underlying pathophysiological process as well as providing vasodilatation.
Current medical treatment for severe pulmonary hypertension is now so effective in those who respond acutely that it is preferable to heart/lung transplantation. It is hoped that calcium channel antagonists with less detrimental effect on myocardial contractility may be developed for use in PPH. At present calcium channel antagonists remain the oral drug of choice as vasodilators in severe PPH. Their benefit in pulmonary hypertension secondary to other conditions such as connective tissue disease has not been established.39
Other vasodilators
Many other vasodilators have been used in PPH, reflecting the fact that none is perfect. These include diazoxide,40captopril,41 ,42 and hydralazine.43 At present calcium channel blockers remain the drug of choice despite their problems. Newer vasodilators such as potassium channel openers may become available in the future but they have not yet been evaluated in pulmonary hypertension. Specific potassium channels are present in pulmonary vascular smooth muscle cells and these may well be the avenue by which hypoxia causes pulmonary vasoconstriction.
Nitric oxide has been successfully used as an acute vasodilator in patients with pulmonary hypertension but at present the need for it to be delivered in a continuous gaseous form means that it is not really an option for long term treatment. Nitric oxide donors are being developed which could be used both orally and by inhalation and this may be the future for these compounds. They are of particular interest because of the known endothelial cell dysfunction in PPH.
Inotropic agents
Since right heart dysfunction is a sequel of severe pulmonary hypertension due to the increased outflow impedance of the right ventricle, it would appear logical to use inotropes once maximum vasodilation has been achieved, particularly if there is concomitant use of calcium channel blockers which are known to be negatively inotropic. No large clinical trials have looked at the use of directly acting inotropes in precapillary pulmonary hypertension, but a recent study using digoxin showed an improvement in cardiac output and a fall in sympathomimetic amines in pulmonary hypertension.44While there are few data on the benefit of inotropic agents, there is considerable evidence that other agents such as β blockers and ACE inhibitors used to treat biventricular heart failure of ischaemic origin may cause deterioration and should not be used. The co-existence of pulmonary hypertension with portal hypertension may be a special case, perhaps because a vasodilator may affect both circulations. There is anecdotal evidence of benefit for isosorbide mononitrate in a patient with pulmonary hypertension associated with portal hypertension due to hepatitis C virus.45 She responded to intravenous prostacyclin and, since isosorbide is commonly used for portal hypertension, was given isosorbide 40 mg twice daily. The second catheterisation six months later showed a beneficial effect on pulmonary haemodynamics.
Prostacyclin
The largest literature on the use of vasodilators in severe pulmonary hypertension relates to prostacyclin and analogues. Prostacyclin is a natural vasodilator produced by the endothelial cells of the circulation. Rubin et al first showed that prostacyclin induces vasodilation in patients with PPH.46 Higenbottam then used it as a bridge to transplantation in a young woman with severe pulmonary hypertension. She was restored from being bed bound to an active life while awaiting transplantation.47 It was not until 1990 that the drug was assessed in a large group of patients by Rubin et al.48 They showed a reduction in pulmonary artery pressure of more than 10 mm Hg in six of 10 patients who completed an eight week study and the benefits of continuing therapy were seen up to 18 months. Continuous intravenous prostacyclin also produced benefits over conventional treatment when used in PPH. Barstet al for the Primary Pulmonary Hypertension Study Group conducted a 12 week prospective trial comparing continuous intravenous prostacyclin with conventional therapy (anticoagulation and calcium antagonists) in 81 patients with severe pulmonary hypertension. Those receiving continuous prostacyclin showed an improvement in exercise tolerance whereas those on conventional treatment showed a fall over 12 weeks. There were also improvements in haemodynamics consistent with the view that improved haemodynamics were responsible for the improvement in exercise tolerance.20 They then looked at the effects of longer term prostacyclin (more than one year) in 27 patients with primary and secondary pulmonary hypertension who had demonstrated a mean reduction of 27% in pulmonary vascular resistance with an intravenous vasodilator.49 The rate of infusion of prostacyclin was increased by, on average, 2.4 ng/kg body weight per minute per month. Twenty six of the 27 patients showed improvements in both symptoms and haemodynamic measurements with a mean fall in pulmonary vascular resistance of 53%. Of interest was the fact that the mean fall in pulmonary artery pressure over the study period was greater than that seen acutely with maximal doses of intravenous adenosine. This suggests that long term prostacyclin has effects other than those of simple vasodilation, presumably causing some reversal of the histological changes in the vessels of the pulmonary circulation. These studies have changed the whole basis of how we view severe pulmonary hypertension. We used to believe that prostacyclin was simply a vasodilator and therefore could only prevent further worsening of the disease. It is now clear that the disease process can be reversed and it is tempting to speculate that the histological changes are also reversed. However, a major hazard with continuous intravenous prostacyclin therapy is that it requires an indwelling Hickman line and pump. The indwelling line carries the risk of infection, catheter clot, catheter breakage, and catheter dislodgement. Patients must be taught scrupulous aseptic technique, how to make up their own drug, and to attach the syringes. Another problem is that, should supply cease for any reason, they can develop life threatening pulmonary hypertension and die. Thus, all patients must be given a back up syringe system. It would clearly be preferable if the drug could be given in other ways and there is evidence that prostacyclin and its analogues can, like nitric oxide, be given by inhalation.50-52
Iloprost has similar effects to prostacyclin when given intravenously53 but, although it has a longer half life, this confers no real advantage over prostacyclin when given intravenously. However, its long half life may be of benefit because it can be given via a nebuliser and a large prospective trial of inhaled iloprost is being considered for later this year. Other analogues with different half lives have been tried by other routes or are under investigation including continuous subcutaneous infusion, but the results have not yet been reported. Clearly, the best route for administering these agents would be orally and oral beraprost, a prostacyclin analogue, has been tried in small groups of Japanese patients but it is not yet available for clinical use.54 ,55
The principal problem with prostacyclin is its cost which, for the average UK patient, is between £40 000 and £60 000 a year. Clearly, this is a major burden for any health authority (health board in Scotland) and thus there is a need to centralise the use of prostacyclin to a limited number of pulmonary vascular units spread around the UK.
Surgical methods
Atrial septal defects (ASD) with left to right shunts are a cause of pulmonary hypertension. However, in situations where severe pre-capillary pulmonary hypertension develops, the creation of an artificial ASD is a possible treatment. There have been no formal clinical trials of atrial septostomy but a number of centres have performed the procedure with evidence of benefit.56-58The rationale for atrial septostomy is that unloading the right heart and improving pre-load to the left heart via the atrial septal defect will improve cardiac output. Oxygen content of arterial blood falls but, as cardiac output rises, oxygen delivery should improve. The treatment is not yet standardised but can be performed by either balloon or blade septostomy. It can be considered in patients with recurrent syncope or right heart failure despite maximum medical therapy when no other therapeutic option exists apart from transplantation. Clearly, this treatment should only be undertaken in centres with experience of septostomy and severe pulmonary hypertension.
Transplantation
Before the improvement in medical treatment for severe pulmonary hypertension, heart/lung or lung transplantation was the standard approach for severe intractable pulmonary hypertension and, in some centres, a large proportion of transplants were for this diagnosis. With over 1000 patients worldwide having received either single lung, double lung, or heart/lung transplantation, there is considerable experience of transplantation for pulmonary hypertension. Surprisingly, there appears to be little difference in the effectiveness of one operation over another although patients undergoing single lung transplantation for pulmonary hypertension have fared slightly less well than those having a single lung transplant for other conditions. A detailed analysis of the costs and benefits of transplantation is outside the scope of this article but at present we can expect a one year survival of 70% and a three year survival of 50–60% in patients with pulmonary hypertension. These figures are slightly worse than those for aggressive vasodilator therapy in “responders”. The principal problems with transplantation are the need for continuous immunosuppression and the development of late complications such as bronchiolitis obliterans. The better results with medical treatment make the timing of referral to a transplant centre more difficult. In general, however, if patients are failing to improve on medical treatment they should be considered for referral for transplantation; they can always be evaluated and referred back to the pulmonary vascular centre if not thought to be sufficiently ill for a transplant at the time of evaluation. There are a number of reviews of strategy regarding the timing of transplantation in pulmonary hypertension but the future of conventional transplantation is bleak because donors are in very short supply.59-61 Despite adequate facilities in most Western countries, the principal rate limiting step is the availability of donor organs. This has prompted some live donations from living relatives but the associated moral and ethical dilemmas are considerable. There is a possibility that animal donors such as the pig may be available in the future once the immunological problems are conquered but, once again, there are ethical issues and fears of cross infection.
Pathobiology of severe pulmonary hypertension
There are many associations and risks for severe intrinsic pulmonary hypertension including congenital shunts, HIV infection,62-64 anorexigens,65-69 portal hypertension,70 autoimmune disease,71-74hypoxia, and there is the familial form of the disease. Despite the large number of associated risks and causes for severe pulmonary hypertension, there is a surprising similarity in the histological change seen in the pulmonary vessels. Preliminary data also suggest surprising similarities in the response to treatment, particularly to intravenous prostacyclin which, whether or not it causes vasodilation when given acutely, may result in a fall in pressures long term which suggests that it is working as an anti-growth factor and reversing the vascular remodelling process. At present no common underlying mechanism has been found to link all these conditions but it is likely that one will be found in the future. One question is the link between vasoconstriction and remodelling. Does vasoconstriction lead to remodelling and does vasodilation lead to de-remodelling? It is tempting to say that this is the case because many vasoconstrictors are also growth factors and many vasodilators are anti-growth factors, allowing us to postulate cell growth–vasomotor coupling.75 To find a common mechanism it is worth looking at the cellular make up of the pulmonary vessels and to consider the possible factors, whether physical or inflammatory, to which they might be exposed and the ways in which they might respond. Firstly, however, we must consider how genetic predisposition might dictate the cellular response to extrinsic factors.
GENETICS OF PULMONARY HYPERTENSION
It is clear that genetic analysis is going to play an important part in elucidating the pathobiology of PPH. The familial form of the disease appears to depend on inheritance of one or perhaps two PPH genes.76 ,77 Approximately 100 families worldwide with familial PPH have been identified including 72 in the USA, eight in the UK, and 10 in Australia. Overall, the incidence of familial pulmonary hypertension is approximately 6%. Inheritance appears to be via vertical transmission, suggesting a single dominant gene. It is not sex linked and is likely to be an autosomal dominant gene with incomplete penetrance. The syndrome can skip generations and having the gene confers only a 10–20% likelihood of developing the disease. The first gene ascribed to PPH (given the code PPH1) has been localised to seven million base pairs at 2q 31–32.78 Genetic anticipation appears to occur with successive generations developing the disease slightly earlier than the preceding generation. Most recently, a family study in the UK has discovered a gene outside the 2q 31–32 region, suggesting heterogeneity of the genetic basis of familial PPH but further details are needed. At present the proteins coded by the PPH gene(s) are unknown.
As well as familial PPH there is also evidence of association particularly with autoimmune diseases suggesting co-inheritance. For example, approximately 10% of patients with PPH have Raynaud's phenomenon and up to 40% of patients with autoimmune disease, particularly the CREST variant of systemic sclerosis, have pulmonary hypertension. No genetic basis for this association has been determined but co-association provides further evidence to suggest an immunogenetic basis for the disease.
PATHOBIOLOGY OF CELLULAR CHANGE IN SEVERE INTRINSIC PULMONARY HYPERTENSION
Changes are seen in each of the three main cell types that make up the pulmonary vessels (for review see Wagenwoort15). In the intima there is intimal thickening and fibrosis. In the media there is increased muscularity leading to the increased thickness of the muscular arteries and also muscularisation of arterioles which do not normally have a muscular coat. There is also increased connective tissue and extracellular matrix, particularly in the larger muscular arteries. In the adventitia there is an increase in matrix protein probably due in part to fibroblast proliferation (fig5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cross section of a pulmonary artery (approximately 125 μm) in a patient with severe pulmonary hypertension showing thickening of all three layers of the vessel. Possible mechanisms for these changes are suggested. 1=intima; 2=media; 3=adrentitia.
Endothelial cells
The endothelial cells form the intima of pulmonary arteries and are usually one layer thick. The endothelium is considered to be the front line in response to haemodynamic changes in the circulation and may also be the concert leader in terms of directing the remodelling process that occurs in response to haemodynamic change, inflammation, injury and circulating agents. The endothelium is a source of vasoconstrictors (endothelin 1 and thromboxane) and vasodilators (prostacyclin and nitric oxide). It is useful to think of the tone of a vessel as being determined by the balance of the vasodilators and vasoconstrictors. The vasoactive agents may also be responsible for cellular remodelling (see above) so that disease of the pulmonary circulation can be considered as an imbalance between vasoconstrictor/growth factors and vasodilator/anti-growth factors. We know, for example, that there is nitric oxide deficiency in pulmonary hypertension induced by appetite suppressants.79 There is impairment of endothelium mediated pulmonary vasodilation in patients with both PPH and systemic sclerosis,80 ,81 suggesting a reduction in vasodilator capacity, an increase in vasoconstrictor capacity, or a change in the balance between the two. There is evidence for all of these. For example, decreased nitric oxide concentrations have been found in the exhaled air of patients with pulmonary hypertension with systemic sclerosis82 and patients with PPH have shown reduced expression of nitric oxide synthase,83 an increase in circulating endothelin 184 and 5-hydroxytryptamine,85 and an imbalance between vasoconstrictors and vasodilators was seen in patients with pulmonary hypertension of different causes.86 Changes in nitric oxide production are thought to be responsible for the abnormal arteriovenous connections seen in patients with cirrhosis of the liver. In these patients the physiological vasodilation of pulmonary capillaries may possibly be due to excess nitric oxide production resulting in right to left shunts and hypoxaemia. In contrast, some patients with cirrhosis, usually those with portal hypertension, develop pulmonary hypertension. The mechanism remains unknown but has been reviewed by Herve et al.70
Endothelin 1 is a powerful pulmonary vasoconstrictor although its role in the physiological control of the pulmonary circulation is unknown. It appears to have little influence on acute vasoconstriction as seen with hypoxic pulmonary vasoconstriction but it may have a role in conditions causing prolonged severe intrinsic pulmonary hypertension where its slow onset of action may be more relevant. There is evidence of increased expression of endothelin 1 in patients with pulmonary hypertension.84
Media
The media is composed of smooth muscle cells which, in patients with PPH, become hypertrophied and the muscularisation then extends into previously non-muscular areas. In addition, there is laying down of extracellular matrix. The changes in the media may be orchestrated from the endothelium or may be due to changes in potassium channels in the cells87 or to increased circulating vasoconstrictors such as serotonin.85 ,88
Adventitia
The adventitia is composed of matrix proteins including collagen and elastin secreted by fibroblasts. There may be fibroblast proliferation and differentiation and an increase in matrix proteins. These changes are likely to be due to physical factors such as stretch and changes in pressure89 or hypoxia.90 ,91
Conclusions
It is clear that, for intrinsic pulmonary vascular hypertension to develop, there must be a complex interaction between various elements. There are genetic factors which may be a single PPH gene (the protein product of which is not yet identified) or co-inheritance of other genes such as those responsible for connective tissue disease. Inheritance of these genes may or may not lead to pulmonary hypertension and it is possible that additional factor(s) such as physical injury (hypoxia, sheer stress, pressure and flow), inflammation (autoimmune disease, HIV disease, local injury (thrombosis)) or circulating toxins (appetite suppressants) are necessary to trigger a process which involves vasoconstriction and cellular remodelling. A complex interplay of vasodilators, vasoconstrictors, growth factors, and anti-growth factors may be necessary to produce the final histological change seen in fig 5, but which factors are present early in the disease and which are a consequence of the disease process is not yet clear. It is likely that, in the next 10 years, the genetic and physiopathological basis underlying severe pulmonary hypertension will be understood. Hopefully, this will lead to a radical change in treatment and a further improvement in morbidity for a devastating set of diseases which often affect young people.
UK Pulmonary Vascular Units
Dr Andrew J Peacock, Scottish Pulmonary Vascular Unit, Western Infirmary, Glasgow G11 6NT Tel: 0141 211 1836 Fax: 0141 211 6334 Professor Tim Higgenbottam, Pulmonary Hypertension Unit, Division of Clinical Sciences, (CSUH Trust), The Medical School, Beech Hill Road, Sheffield S10 2RX Tel: 0114 271 2132 Fax: 0114 271 3685/1711 Dr Paul Corris, Department of Cardiology, Freeman Hospital, High Heaton, Newcastle upon Tyne NE7 7DN Tel: 0191 284 3111 Fax: 0191 223 1175/1152 Dr Simon Gibbs, Department of Clinical Cardiology, Charing Cross Hospital, London W6 8RF Tel: 0181 743 2030 Fax: 0181 746 8182 Professor Carol Black, Department of Rheumatology, Royal Free Hospital, London NW3 2QG Tel: 0171 794 0500 Fax: 0171 435 0143 Professor Alyn Morice, Academic Department of Medicine, Castlehill Hospital, Cottingham HU16 5JQTel: 01482 875875 Fax: 01482 624068 Dr Keith McNeil, Transplant Unit, Papworth Hospital, Papworth Everard, Cambridge CB3 8RE Tel: 01480 830541 Fax: 01480 364610
References
Linked Articles
- Correction