Article Text

Statistics from Altmetric.com
Streptococcus pneumoniae was identified as a major respiratory pathogen shortly after its isolation in 1881.1 Despite a century of intensive study, and antibiotics which readily kill the organism, respiratory tract infections caused by the pneumococcus remain a formidable problem.S pneumoniae is the commonest cause of community acquired pneumonia, accounting for up to 70% of cases in hospital.2 ,3 Pneumococcal pneumonia is associated with bacteraemia more frequently than other bacterial pneumonias4 and mortality from bacteraemic pneumococcal pneumonia during the first few days of hospitalisation has changed little since the pre-antibiotic era.5 ,6 There is also evidence, from the UK and other countries, that the number of cases of pneumococcal bacteraemia is rising.7 ,8 In the third world five million children under the age of five die each year from acute lower respiratory tract infections in which S pneumoniae is probably the primary agent,9 and patients with HIV infection and AIDS also have a high risk of pneumococcal pneumonia and bacteraemia.10 Furthermore, the worldwide increase in penicillin resistance among pneumococci11-15 and the limited use of the pneumococcal vaccine16 suggest that morbidity and mortality from pneumococcal disease may increase.
In recent years there has been an increased understanding of the interactions between the pneumococcus and the host, both in terms of how the virulence factors of the organism contribute to the pathogenesis of pneumonia and how the host’s response to infection can be harmful as well as protective. The role of cytokines in pneumococcal pneumonia, the detailed behaviour of neutrophils in the disease, and the mechanisms by which the pneumococcus attaches to the host both during nasopharyngeal colonisation and during invasive disease have all been the subject of recent investigation. Whilst antibiotics and supportive care are likely to remain the keystone of the treatment of established infection, these areas of research offer the best prospects for reducing the high incidence of the disease and the high mortality during the first few days following hospital admission.17
This review will therefore concentrate on the pathogenesis of pneumococcal pneumonia, with emphasis on recent advances at the cellular and molecular level. For descriptions of the epidemiology, diagnosis and management of the disease, and for an up to date evaluation of the effectiveness of the current pneumococcal vaccine, readers are referred to other recent reviews which cover these subjects comprehensively.18-20
The organism: virulence factors and antibiotic resistance
Although S pneumoniae exists in encapsulated and unencapsulated forms, only encapsulated strains have been isolated from clinical material. The importance of the capsule in pneumococcal virulence was first established by enzymatic removal of the capsule,21 and has recently been confirmed using genetically engineered pneumococci which differ only in capsular type. The virulence of the mutants in relation to the parental strains was determined mainly, though not entirely, by the capsular type.22
However, the capsule itself is not toxic.23 Composed of one of 90 serologically distinct polysaccharides,24 ,25the virulence of the capsule lies mainly in its antiphagocytic properties (fig 1). The level of virulence is determined more by the chemical nature of the capsule then by its size,26 and the production of serotype specific protective antibody in response to capsular polysaccharide is the basis of the current anti-pneumococcal vaccine. Geographical, temporal, and age differences in the distribution of the 90 different serotypes24 ,25 and the ability of S pneumoniae to transfer cassettes of capsular genes from one strain to another leading to a change in capsule specificity27 ,28 all have potential implications for vaccine strategy.25 ,29 The current 23 valent vaccine includes the serotypes that cause 88% of the bacteraemic infections in the USA30 and 96% of those in the UK.18

Schematic figure of the known virulence factors of Streptococcus pneumoniae including their main functions and cellular location. Only factors with proven virulence are included. For other putative virulence factors, see text.
In contrast to the capsule, the cell wall is a potent inducer of inflammation,23 probably via the activation of complement31 and the induction of cytokines.32 ,33 The active component is cell wall polysaccharide, a complex teichoic acid which—very unusually among bacteria—contains phosphorylcholine.34 As discussed later, phosphorylcholine provides a site of attachment to activated endothelial cells during the course of invasive disease. Antibody against cell wall polysaccharide or phosphorylcholine is protective against pneumococcal challenge, though the protective effect is substantially weaker than that of antibody to capsular polysaccharide.35
In addition to surface polysaccharides, the pneumococcus contains a number of proteins that have been shown to contribute to virulence (fig1).35-39 These comprise pneumolysin, an intracellular toxin which is released only when the cell wall undergoes lysis; autolysin, the enzyme which is responsible for cell wall lysis; and pneumococcal surface protein A (PspA), a protein on the cell surface which is highly immunogenic in mice.39 Other pneumococcal products which may contribute to the pathogenicity of the organism, but whose role in virulence has not been established, include neuraminidase,40 ,41 hyaluronidase,42 a neutrophil elastase inhibitor,43 various proteases including an enzyme which cleaves human IgA1,44 ,45 an inhibitor of the respiratory burst of neutrophils,46 ,47 and a number of putative protein adhesins.35 ,48-52
The most extensively studied of the protein virulence factors is pneumolysin, a toxin which lyses cholesterol-containing cell membranes and activates complement. Pneumolysin has a variety of detrimental effects on host cells and functions in vitro,36 and causes a severe lobar pneumonia when injected into the apical bronchus of rats.53 More direct evidence for its role in pathogenicity comes from the demonstration that laboratory mutated strains of pneumococci deficient in pneumolysin have reduced virulence compared with wild type organisms36 ,54-56 and that immunisation with pneumolysin or its toxoid protects mice against subsequent challenge with virulent pneumococci.36 The cytolytic and complement activating properties of pneumolysin have recently been mapped to separate regions of the molecule. Using strains of pneumococci with mutations for each of these molecular sites, it has been shown that both sites contribute to the early pathogenesis of pneumococcal pneumonia at different stages of infection and by different mechanisms.56-58 Similar studies with antisera and genetically engineered mutants have demonstrated the contributions of autolysin35 ,59 and PspA39 to pathogenicity.
Interest in pneumococcal proteins lies not only in their pathogenicity but also in the fact that, as proteins, they are T dependent antigens and have the potential to be used to improve pneumococcal vaccines. The current vaccine is based on capsular polysaccharides which, as T independent antigens—that is, they can elicit an antibody response by stimulating B lymphocytes directly without the help of T cells—have two disadvantages: very poor immunogenicity in children under the age of two years and the lack of a memory booster response on rechallenge with antigen. By conjugating the polysaccharide antigens to a protein, however, they can be converted into a T dependent form which does not have these drawbacks.17 ,35 ,38 Although there is no immunological necessity for the carrier protein to be from the pneumococcus, a pneumococcal protein may have the advantage of conferring species-specific immunity. One potential disadvantage of the new conjugate vaccines, however, is that only a limited number of serotypes may be included. To determine whether this outweighs the potential advantages it will be necessary to follow both the incidence of pneumococcal disease and the serotype distribution of pneumococci after introduction of the conjugate vaccines.25
Another group of clinically relevant proteins are the transcarboxypeptidases in the cell wall which also bind penicillin (penicillin binding proteins).11 ,38 Alterations to the penicillin binding properties of these proteins are brought about by transfer of portions of the genes for the penicillin binding proteins from other streptococcal species resulting in mosaic genes, and can occur without affecting the cell wall building functions of the enzymes. Because only portions of the genes are transferred, and because there are a range of penicillin binding proteins which can be modified in a stepwise manner, the level of resistance to penicillin can vary considerably.11
This graded nature of penicillin resistance has direct relevance to clinical practice, for most “resistant” species ofS pneumoniae isolated from clinical samples show only intermediate resistance (minimum inhibitory concentration (MIC) 0.12–1.0 μg/ml) to penicillin. These MICs can easily be exceeded in the lungs by intravenous treatment with high dose penicillin, and Pallares et al 12 have recently shown, that even in Spain where the instance of pneumococcal resistance to penicillin is high, the administration of penicillin G or ampicillin in high doses intravenously is still an effective treatment for pneumococcal pneumonia. These authors recommend an alternative—a third generation cephalosporin—only when community acquired pneumonia is life threatening or when risk factors are present for high level penicillin resistance (MIC ⩾2.0 μg/ml). It should be noted that the same principles have not been shown to apply to the treatment of erythromycin resistant S pneumoniae. Although some of the new macrolides have better lung penetration than erythromycin, clinical studies with these agents have not been performed in patients with erythromycin resistant pneumococcal pneumonia, and in such patients it is recommended that non-macrolide agents be used.15 ,19
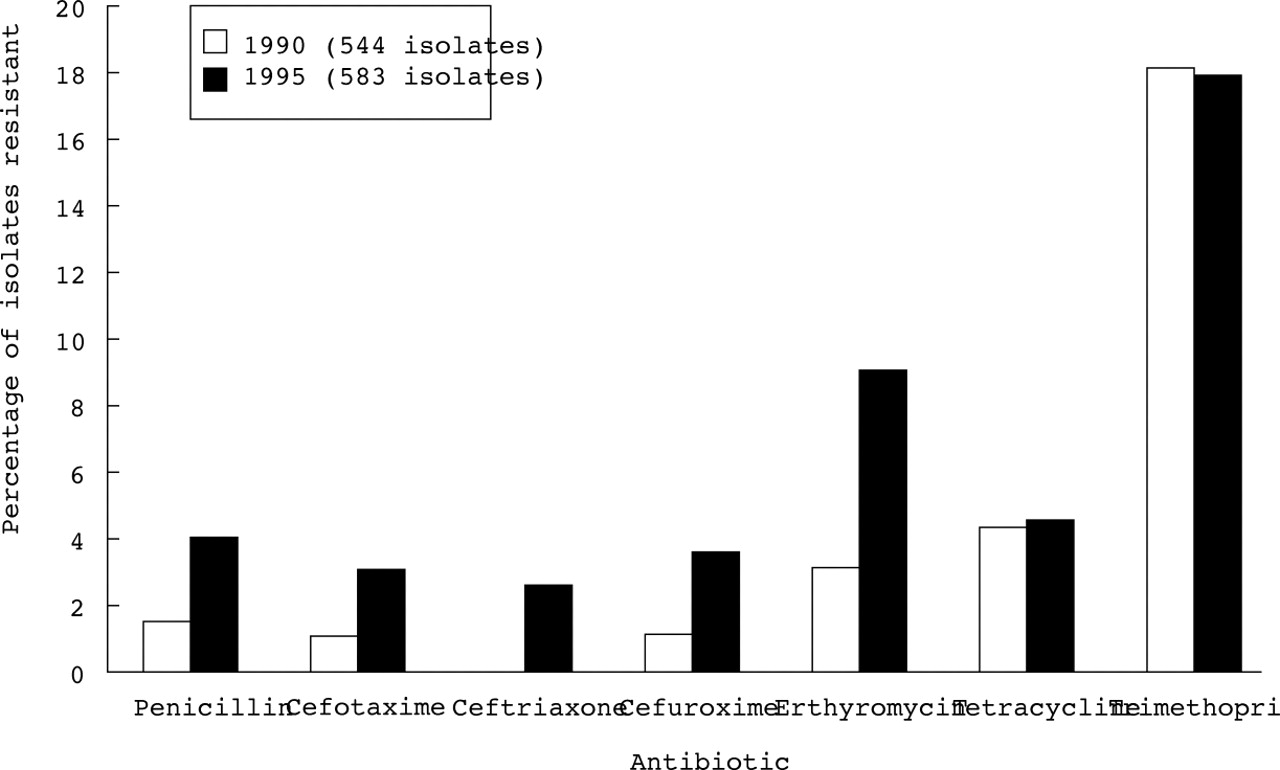
Resistance to penicillin in S pneumoniae is increasing throughout the world.11-15 The problem is particularly common in Spain, Eastern Europe, South Africa, South America, New Guinea, and Korea where resistance up to 30–50% is commonly reported.11 However, even in countries with generally low numbers of resistant organisms, including the UK, the proportion of S pneumoniae resistant to penicillin is steadily increasing, along with resistance to erythromycin and other antibiotics (fig 2).14 ,15 Most of the resistant strains belong to a small number of serotypes (6, 14, 19, and 23) which are prevalent in young children and which are being included in the new conjugate vaccines.14

Nasopharyngeal colonisation
The initial step in the pathogenesis of pneumococcal infection is the attachment of the organism to the mucus and cells of the nasopharynx.60-62 Acquisition of the causative strain usually occurs during the month preceding clinical infection, and studies in closed communities,63 including recent observations on the spread of penicillin resistant pneumonia in children attending nurseries,64 have shown that rates of pneumococcal disease depend on the frequency with which invasive serotypes are carried by healthy individuals.
However, colonisation frequently occurs without the development of disease. Colonisation can occur within hours of birth and by the 12th postnasal day the carrier rate is similar to that of the babies’ mothers.63 Carriage rates are highest in pre-school children, whilst rates amongst adults depend on the likelihood of contact with children.64 Up to four different serotypes can be found at one time usually for periods of several weeks or months.65-67 Recent studies during two outbreaks of pneumococcal pneumonia in military camps have shown that asymptomatic nasopharyngeal colonisation with S pneumoniae frequently results in the production of circulating type specific antibody at levels which confer protection from pneumonia against that serotype.68 It therefore appears that, although aspiration of colonising organisms during the first few weeks of colonisation may lead to pneumonia, after that time most healthy adults are likely to be protected.
At the molecular level adherence of S pneumoniae to nasopharyngeal cells probably occurs through interaction with a specific disaccharide (N-acetyl-d-glucosamine β1–3 galactose, GlcNAcβ1–3Cal) which forms part of a glycolipid receptor on the epithelial cell surface.62 The pneumococcal ligand which binds to this specific disaccharide has not yet been identified, but it has been observed that pneumococci will only bind to nasopharyngeal cells and to this receptor when they are in a particular morphological phase.69 ,70 It is hoped that the biochemical basis of this morphological phase change will be elucidated and help to clarify the pneumococcal factors which favour adherence.
Transition from colonisation to pneumonia and invasive disease
The factors which permit pneumococci to spread beyond the nasopharynx are poorly characterised and are likely to vary depending on the virulence of the organism, the state of the host’s defences, and the existence of preceding viral infection. Spread to the lungs probably occurs by aspiration71 which is aided by impairment of the cough reflex, by increased production of mucus (in which pneumococci also replicate61), and by impairment of the mucociliary escalator. Whilst all of these can be caused by host related disorders, the pneumococcus itself can contribute by causing impaired ciliary activity, and by pneumolysin dependent disruption of the epithelial type junctions which are essential for the production of normal mucus.54 ,72 Both influenza virus and adenovirus enhance in vitro adherence of S pneumoniaeto respiratory tract epithelial cells.60
Recent studies have shown that S pneumoniaecan adhere to human bronchial epithelial cells.73However, the molecular basis of this interaction is not known, nor is its pathogenetic significance, either in pneumonia or in acute or chronic bronchitis. In healthy individuals the bronchi are sterile, but in patients with chronic bronchitis the organism can be present in the lower respiratory tract even in between exacerbations.74To date, our understanding of this form of colonisation is poor, as is the relationship between this and acute exacerbations of chronic obstructive pulmonary disease (COPD). Attempts to evaluate the role ofS pneumoniae have been hampered by the presence of colonising organisms in the nasopharynx and lower respiratory tract and the lack of a standard definition of an acute exacerbation of COPD.74
Within the alveoli pneumococci initially adhere to the alveolar wall, probably preferentially to alveolar type II cells.75 They also spread very rapidly to the blood, suggesting an aggressive capacity to cross the vascular endothelial cells.71Attachment of S pneumoniae to resting type II lung cells and vascular endothelial cells is mediated by host cell glycoconjugate receptors with specific disaccharides (N-acetyl-d-glucosamine β1–4 galactose, GalNAC β1–4 Gal, or N-acetyl galactosamine β1–3 galactose, GalNAc β1–4 Gal) as the minimum receptor subunits. These galactose based disaccharides are similar to, but distinct from, the glucosamine based disaccharide receptor on nasopharyngeal epithelial cells.75Pneumococcal binding to these glycoconjugates is mediated both by phosphorylcholine in the cell wall76 and by pneumococcal proteins. By using libraries of genetic mutants ofS pneumoniae with defects in specific proteins,77 a number of putative protein adhesins have been identified.35 ,48-52 However, it is likely that many of these proteins, some of which function as enzymes, act indirectly in this role.48
When these lung and endothelial cells are activated, however, there is evidence that the pneumococcus shifts its target to a new receptor, that for platelet activating factor (PAF).
Cundell et al 78 found that cytokine activation of endothelial cells and lung cells was associated with a marked increase in expression of PAF receptor compared with resting cells, and that pneumococci adhered to the receptor via phosphorylcholine, a determinant which it shares with PAF, with both increased adherence of pneumococci to the cells and increased invasion of the cells. Only virulent pneumococci engaged the PAF receptor, and the sequence of events could be arrested in vitro and in vivo by pretreatment with PAF receptor-specific antagonists. The authors suggested that this may be a critical and potentially reversible step in the transition from asymptomatic colonisation to invasive disease.
Development and resolution of pneumonia
The earliest histological abnormality in pneumococcal pneumonia is engorgement of the alveoli with proteinaceous fluid and red blood cells67 due to damage and increased permeability of the alveolar-capillary barrier. Fibrin also forms, due to the induction of procoagulant activity on human endothelial cells byS pneumoniae, reducing the blood supply to the affected area and also permitting neutrophils to trap the pneumococci against the surface of pulmonary cells.67 ,79A range of studies involving animal models of pneumonia and in vitro studies with monolayers of type I alveolar epithelial cells and vascular endothelial cells have shown that these cells, and the tight junctions between them, can be damaged either by pneumococcal cell wall products32 or by pneumolysin,57 ,58 though there is evidence that, with cell wall products, the changes are mediated by tumour necrosis factor (TNF) from the host since the cytopathic effect on endothelial cells can be abrogated by anti-TNF antibodies.23 ,32 These events do not require the presence of neutrophils80 and, as with other aspects of the inflammatory process, there is no evidence that the pneumococcal capsule plays a part.23
Clearance of pneumococci from the alveoli by phagocytosis is dependent on complement31 ,65 and facilitated by anticapsular antibody,35 ,65 both of which function opsonically. Resident alveolar macrophages play a phagocytic role in the early stage of infection81 but after 1–2 hours they are joined and quickly outnumbered by the more efficient polymorphonuclear leucocytes (PMNs).79 The PMNs are sequestered into capillaries throughout the lung but migrate into alveoli only at the site of infection.79 The chemotaxins involved include the complement factor C-5a82 which is released by direct activation of complement by pneumococcal cell wall teichoic acid,31 and both interleukin 8 (IL-8)83 and leukotriene B484 released from alveolar macrophages. However, the molecular pathway by which neutrophils migrate from the vascular to the alveolar space has not been established. Unlike many processes involving sequestration and migration of neutrophils, the process does not depend on selectins85 ,86 and it is also probably independent of CD11-CD18 adhesion molecules,87 ,88 even though there is evidence of upregulation of some of these adhesion molecules in community acquired pneumonia.89 This lack of dependence on selectins and CD11-CD18 appears partly to reflect anatomical differences between the pulmonary and the systemic microvasculature but also probably differences between pneumococci and other organisms.68 ,86
The increased demand for PMNs in the blood and lungs is met by increased release of new cells from the bone marrow.79These cells are immature,90 having had a shorter transit time in the bone marrow91 and, as such, are larger and less deformable than mature PMNs. It has been observed that only a small proportion of these immature cells migrate into the alveoli, leaving an excess in the pulmonary vascular space.90 Since immature PMNs, when stimulated, have a greater potential to release oxygen radicals and proteolytic enzymes than mature cells, it has been suggested that pneumococcal bacteraemia may result in intravascular activation of these cells, leading to lung injury, and possibly providing an explanation for the high mortality associated with bacteraemia in pneumococcal pneumonia.90
However, one of the remarkable features of pneumococcal pneumonia is that the lungs of patients who recover from the disease almost invariably return to normal, irrespective of the severity of the systemic or pulmonary condition when the disease was at its peak. Whilst this has been recognised since early descriptions of the disease,92 ,93 it is only recently that the details of possible contributory mechanisms have emerged. It has been found, for example, that alveolar macrophages from patients with community acquired pneumonia are hyporesponsive to stimulation, with less release of pro-inflammatory cytokines than either circulating monocytes from the same patients or from alveolar macrophages from healthy control subjects.94 This is in keeping with the general tendency of alveolar macrophages to reduce or dampen down the immune response within the alveoli.95 It has also been observed that viable S pneumoniae in suspension inhibit the release of reactive oxygen species from PMNs, in contrast toStaphylococcus aureus andKlebsiella pneumoniae which each stimulate PMNs to release increased amounts of extracellular reactive oxygen species.46 ,47 This inhibition of the neutrophil oxidative burst by S pneumoniae may help to explain why the pneumococcus rarely causes permanent lung damage whereas pneumonia caused by the other two organisms is often associated with abscess formation and fibrosis. Furthermore, in experimental pneumococcal pneumonia it has been shown that PMN recruitment to the lungs is complete within 24 hours,92 ,96 that PMN activity as measured by the uptake of a radiolabelled glucose analogue ceases within 48 hours,97 and that, before PMNs are cleared by macrophages, they readily undergo apoptosis—that is, death in a programmed or “physiological” fashion rather than releasing their potentially toxic products on death.92 ,93 All of these aspects of neutrophil activity will reduce the chances of lung damage.
Bacteraemia, systemic antibody and the role of the spleen
The risks of bacteraemia and death from pneumococcal pneumonia are greatly reduced by the presence of type specific anticapsular antibody in serum.35 Generation of the antibody was shown by early investigators to herald recovery from invasive pneumococcal disease, and the first effective treatment for pneumococcal pneumonia was provided by passive immunisation with specific antisera.1Antibodies to other pneumococcal components such as cell wall polysaccharide, pneumolysin, PspA, and other proteins are also produced in the course of colonisation or infection but their protective capacity is less.35
However, in the course of pneumococcal infection, antibody is produced late in the sequence of host defences, usually on the 5th or 6th day of disease,98 and not all studies have suggested that this defence is necessarily adequate or the important one. For example, opsonophagocytosis of S pneumoniae can occur in vitro in the absence of specific antibody,99 and in vivo recovery from pneumococcal pneumonia not treated with antibiotics can occur in the pre-antibody phase.98 Furthermore, pneumococcal bacteraemia has been observed despite high levels of preformed anticapsular antibody and with either a fall or no change in the convalescent sera.63 ,64 Although the accuracy of anticapsular antibody levels has been questioned in recent years because of the co-detection of antibodies to cell wall polysaccharide in many immunoassays,100 this is unlikely to provide a full explanation for the lack of a relationship between antibody levels and clinical disease in these studies.
In the absence of type specific antibody the clearance of pneumococci has been shown to rely on the reticuloendothelial system, with the macrophages of the spleen playing a greater relative role than those of the liver.101 This, as well as the spleen’s role in enhancing antibody production,102 presumably accounts for the increased risk of mortality from pneumococcal bacteraemia in asplenic and functionally asplenic patients. In an experimental model of pneumococcal pneumonia the survival rate of splenectomised mice after an aerosol challenge of S pneumoniaewas improved more than threefold by administration of granulocyte colony stimulating factor (G-CSF).103 The improvement occurred when G-CSF was administered from 24 hours before to three days after the challenge and was associated with a rise in the number of circulating neutrophils. The therapeutic implications of this are still unknown.104
Role of cytokines
The molecular and cellular events which make up the host’s response to infection are regulated by cytokines, a network of low molecular weight polypeptides secreted by many cells including the alveolar macrophage. However, whilst cytokines can act as beneficial chemical mediators in the course of inflammatory and immunological processes, they can also be harmful.105 ,106 Recent studies in patients with (mainly Gram negative) sepsis and/or the adult respiratory distress syndrome (ARDS) suggest that the net effects depend on the balance between pro-inflammatory cytokines such as TNF-α and IL-1 and anti-inflammatory cytokines such as IL-10, and that disturbance of this balance can be associated with a poor prognosis.107 ,108 The question which arises is whether different cytokine responses account for different clinical outcomes in pneumococcal disease, and specifically whether cytokine imbalance can account for the currently unexplained systemic upset of pneumococcal pneumonia and the many deaths which are unexplained by respiratory failure.
Both clinical and experimental studies have shown that there is a brisk local cytokine response to inhaled S pneumoniae. In two studies of unilateral community acquired pneumonia, for example, one in which TNF-α, IL-1 and IL-6 levels were measured94 and one involving measurement of IL-8,83 the cytokine response was greater in the involved lung than in the paired non-involved lung and, with the exception of IL-6, there was no rise in the circulating levels of these cytokines. Similarly, in an animal model in which mice became ill 16–24 hours after nasal inoculation of S pneumoniae and died of bacteraemic pneumococcal pneumonia within five days, TNF-α, IL-6, and IL-10 levels in the lungs all rose within 12 hours and remained high at 72 hours, but IL-6 was the only cytokine of the three measured which was significantly raised in the blood.109-111 Tasaka et alhave also recently shown increased pulmonary expression of the gene for interferon γ (IFN-γ) early in the course of experimental pneumococcal pneumonia.112
However, evidence of the cytokine response is not confined to the lung. In addition to IL-6 being raised in the serum, circulating levels of G-CSF are raised during the acute phase of respiratory tract infections.113 Nevertheless, G-CSF, which probably contributes to the increased release of neutrophils from the bone marrow,90 is likely to originate in the lung during pneumonia for Tazi et al 114found that alveolar macrophages from patients with pneumonia produce G-CSF spontaneously, in contrast to alveolar macrophages from healthy subjects which produce G-CSF only when stimulated.
More direct evidence that the cytokines contribute to host defence against S pneumoniae has come from passive immunisation experiments with monoclonal antibodies against the cytokines (fig 3) and studies with “knock out” mice which have genes for individual cytokines deleted. In a murine model of pneumococcal pneumonia, for example, intranasal administration of IL-10 in combination with S pneumoniae resulted in decreased lung concentrations of TNF-α and IFN-γ, increased bacterial counts in lungs and blood, and early lethality. Conversely, pretreatment of mice with an anti-IL-10 antibody was associated with increased lung levels of TNF-α and IFN-γ, reduced bacterial counts in lungs and plasma 40 hours after the inoculation, and prolonged survival.109 In another series of experiments mice passively immunised against the pro-inflammatory cytokine TNF-α had increased numbers of S pneumoniae isolated from the lungs and died earlier than non-treated mice.110Using the same model of pneumococcal pneumonia, mice deficient in the gene for IL-6 had increased lung levels both of the pro-inflammatory cytokines TNF-α, IL-1β and IFN-γ and of the anti-inflammatory cytokine IL-10, increased numbers of S pneumoniae in the lungs, and reduced survival compared with controls, indicating a net protective effect of IL-6.111Taken together, along with similar cytokine studies in animal models of pneumonia involving other organisms,104 ,115-117 these results suggest that the compartmentalised cytokine response of pneumonia contributes to host defence, and that interference with the cytokine network in such a way that pulmonary inflammation is reduced is likely to be harmful to the host.

{kind=link}
{kind=link}
{kind=link}
Effects of monoclonal antibodies against (A) the pro-inflammatory cytokine TNF-α and (B) the anti-inflammatory cytokine IL-10 on survival in a murine model of pneumococcal pneumonia. Lower numbers of S pneumoniae were used in the experiments with IL-10. In both series of experiments death of the mice corresponded with the number of surviving pneumococci isolated from blood and lungs. Modified from references 109 and 110.
The relationship between cytokine profiles and clinical outcome in pneumonia remains unclear.118-123 For example, in two studies in which TNF-α was measured in serum there was no relationship with patient outcome118 ,119 whilst in another study patients judged to be critically ill with pneumonia had higher TNF-α levels than patients with pneumonia who were not critically ill.120 The preliminary results of another study, confined to patients with community acquired pneumonia, found that IL-10 levels were significantly more likely to be detectable in patients with systemic inflammatory response syndrome than in patients who did not have this syndrome.121 In two other studies, mainly concerned with ARDS but also including patients with pneumonia as a control group, plasma levels of IL-6 and TNF-α in pneumonia correlated with the degree of lung injury, and there was also a significant relationship between plasma levels of the cytokines and death rate in the study, though this was only significant when patients with ARDS and with pneumonia were grouped together.122 ,123
In view of the complexity of the cytokine network and the large number of variables involved in clinical studies, it is perhaps not surprising that the clinical picture remains unclear. For example, the timing of the measurements in relation to the onset of infection, the presence of cytokine binding proteins, and the technical problem of ex vivo release of cytokines from clinical material can all make interpretation difficult and may account for differences in results between the studies.124 ,125 Furthermore, only a small number of cytokines have been studied to date and in only a small number of patients. Also, studies relating cytokine profiles to clinical outcome have largely involved cytokine measurements in blood even though the primary immune response is known to be in the lung.83 ,94It should also be noted that S pneumoniaewas cultured in only a minority of patients in some studies, and that in other studies patients with pneumococcal pneumonia were grouped with patients who had other pulmonary conditions. It is possible that different infecting organisms and different pulmonary disorders will be associated with different cytokine profiles.126Nevertheless, despite the difficulties involved in studies of this kind, clinical research in this area will be needed both to confirm hypotheses arising from experimental studies and to identify whether there are clinically important differences between individuals in cytokine production, as suggested by recent studies in meningococcal disease.127
Conclusion
The interactions between the pneumococcus and the host are complex. However, the pathogenesis of pneumococcal pneumonia is beginning to be understood at the molecular level, and recent studies have opened the door to the prospect of novel approaches to the prevention and treatment of the condition.
In the foreseeable future conjugate vaccines with pneumococcal proteins are likely to be the most important practical development againstS pneumoniae. It is expected that they will provide the first reliable method of protecting children from pneumococcal pneumonia, and possibly the first protection againstS pneumoniae that is independent of capsular serotype.
In established pneumococcal pneumonia and bacteraemia there is increasing evidence that cytokine imbalance plays an important role in determining outcome. Although progress is limited by the complexity of the cytokine network and the lack of reliable biological markers which can identify the patients most likely to gain potential benefit from cytokine intervention,128 studies in animal models have shown that beneficial effects can be achieved by immunomodulation of this kind, and it is in this area that future research is likely to be targeted.
Acknowledgments
The author thanks Mrs Susan Allsopp, Mrs Katharine Bale and Dr Adrian Kendrick for help in the preparation of this manuscript.