Article Text

Statistics from Altmetric.com
Acute and chronic alveolar and/or bronchial inflammation is thought to be central to the pathogenesis of many lung disorders such as asthma, chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome (ARDS), and idiopathic pulmonary fibrosis (IPF). The site and specific characteristics of the inflammatory responses may be different in each of these diseases, but all are characterised by the recruitment to the lungs and activation of immune and inflammatory cells. These activated cells produce cytokines, oxidants and many other mediators which are involved in inflammation.1 ,2 Recent data indicate that, in addition, airway epithelial cells are able to act as immune effector cells by secreting pro-inflammatory mediators, oxidants, and cytokines.3 Once triggered, an inappropriate chronic inflammatory response persists in these conditions and is presumed to result in lung injury. The intracellular molecular mechanisms in response to environmental signals, leading to increased gene expression and biosynthesis of proinflammatory mediators by airspace inflammatory and epithelial cells, are of considerable current interest. It is now recognised that there are gene specific factors which regulate the transcription of particular genes by affinity binding to specific recognition motifs, which are usually located in the upstream (5′) promoter region of the gene. These factors, which are usually located in the cytosol, can translocate to the cell nucleus and, by binding to specific consensus sites, can upregulate the rate of transcription of the gene and therefore increase the formation of messenger RNA (mRNA) and the protein of an inflammatory mediator. Some transcription factors are cell specific, but others are ubiquitous. Their activity can be modulated by environmental signals and they may play a key role in immune and inflammatory responses. The transcription factors about which there is most information in immune and inflammatory responses are nuclear factor-kappa (κ)B (NF-κB) and activator protein 1 (AP-1).4 Other transcription factors include the nuclear factor for the IL-1 responsive element in the IL-6 gene (NF-IL6), nuclear factor of activated T cells (NFAT), glucocorticoid receptors (GR), cyclic AMP response element binding proteins (CREB), guanine-adenine and thymine-adenine repeats (GATA), E26 transformation specific (ets), and the signal transducers and activators of transcription (STAT) families of transcription factors. The binding sites for these transcription factors are located in the promoter regions of a large variety of genes which are considered to be involved in lung inflammation.5-7 In addition, there are several nuclear receptor co-activators which facilitate DNA binding of transcription factors,8 notably the CREB-binding protein (CBP), adenovirus E1A-associated protein p300/CBP-associated factor (p/CAF), and the steroid receptor co-activator-1 (SRC-1), which interact with basal transcription factors and transcription machinery and are of particular importance in the transactivation and regulation of gene transcription involved in the immune and inflammatory responses. Understanding the function and regulation of basal transcription factors is fundamental to the study of lung inflammation and may provide novel therapeutic strategies for a number of inflammatory lung diseases. In this review we will focus on recent progress in our understanding of the role of the NF-κB, AP-1, NF-IL6, and NFAT transcription factors in the pathogenesis of inflammatory lung diseases and discuss synergistic interactions between GR proteins, other families of transcription factors, and nuclear receptor co-activators which lead to specific gene regulation. We will also review the current concepts of pharmacological intervention to modulate the activation of these transcription factors.
NF-κB
REGULATION OF NF-κB
NF-κB was first identified as a nuclear factor that binds the decameric DNA sequence 5′-GGGACTTTCC-3′ within the intronic immunoglobulin kappa light chain enhancer in mature B cells.9 Binding of NF-κB to this DNA sequence is responsible for the inducible activity of the enhancer element present in the immunoglobulin gene. NF-κB is a member of the Rel family of proteins, a novel family of ubiquitous transcription factors sharing a common structural motif for DNA binding and dimerisation.10 The Rel family of transcription factors can be defined as a group of proteins that share sequence homology over a 300 amino acid region termed the NF-κB/Rel domain. These proteins can exist either as homodimers or heterodimers, each with a specific affinity for different decamer binding sites fitting the κB motif.10 Several different NF-κB proteins have been characterised.10 The classical activated form of NF-κB is a heterodimer which usually consists of two proteins, a 65 kD polypeptide (p65) subunit (also referred to as Rel A) and a 50 kD polypeptide (p50) subunit. Other subunits, such as p105, a p50 precursor (NF-κB1), p100, a precursor of p52 (NF-κB2), c-Rel and Rel B are present in different forms of NF-κB. These subunits form various homodimers and heterodimers which are likely to have different affinities for DNA and different transactivation potentials of the complexes required to activate different sets of specific genes. In unstimulated cells NF-κB is found in the cytoplasm as an inactive non-DNA binding form, associated with an inhibitor protein called inhibitory κB (IκB) which masks the nuclear translocation signal and so prevents NF-κB from entering the nucleus (fig1).10 Upon cell stimulation—for example, by cytokines—specific kinases phosphorylate the IκB-α (IκB kinase complex) and the p105/p65 complex leading to their ubiquitination (transfer of the ubiquitin molecule).11 Ubiquitination of p105 results in an increase in the rate at which p105 is processed to p50, whereas ubiquitination of IκB-α serves as a signal to the proteasome complex (proteolytic enzymes) to degrade rapidly and completely the IκB unit in the cytoplasm.12 This critical release of NF-κB (p65/p50) from IκB results in the translocation of active p65/p50 into the nucleus where it binds to specific motifs in the promoter regions of target genes. The p50 subunit within the p50/p65 heterodimer greatly facilitates DNA binding, whereas the p65 subunit is required for adequate transactivation.10 A p50/p50 homodimer also binds to κB sites but inhibits, rather than triggers, transcription.10The importance of NF-κB subunits is highlighted by the fact that transgenic animals, which do not possess the genes coding for the p50 or Rel B components of NF-κB, have defects in both immune and inflammatory responses.13

Model for the mechanism of NF-κB and AP-1 activation. Activation of NF-κB involves the phosphorylation, ubiquitination, and subsequent proteolytic degradation of the inhibitory protein IκB. Free NF-κB then translocates into the nucleus and binds with its κB consensus sites. Antioxidants such as intracellular glutathione and thioredoxin can inhibit NF-κB activation. Similarly, AP-1 either c-jun/c-jun (homodimer) or c-fos/c-jun (heterodimer) migrates into the nucleus and binds with its TRE consensus regions. Activation of NF-κB/AP-1 leads to gene expression of inflammatory proteins.
ACTIVATORS OF NF-κB
NF-κB activation represents the terminal step in a signal transduction pathway leading from the cell surface to the nucleus. A wide variety of extracellular stimuli trigger the activation of NF-κB (table 1) including pro-inflammatory cytokines,5activators of PKC,10 allergens such as Der p 1,14 and bacterial and viral products.15-17It may be through NF-κB activation that rhinovirus and influenza virus can trigger acute exacerbations of asthma. Experimental infection with rhinovirus activates NF-κB and stimulates the secretion of interleukin 6 (IL-6) in nasal epithelial cells.15
Activators of NF-κB
A conserved cysteine residue has been shown to be critical for both optimal DNA binding and redox regulation of NF-κB protein.18 Reactive oxygen intermediates (ROIs) have been recognised as important inducers of gene expression via NF-κB and AP-1.19 Many of the agents that activate NF-κB, such as TNF-α, IL-1β, lipopolysaccharide (LPS), and ultraviolet (UV) irradiation, also cause an increase in the cellular production of ROIs by mitochondria.20 ROIs have been directly implicated as second messengers in the activation of NF-κB, based upon the ability of oxidants to activate NF-κB by oxidation of its cysteine-SH group or by ubiquitination and proteolysis.21 In addition, hyperoxia,22 ozone,23 and hydrogen peroxide (H2O2),24 iron catalysed lipid peroxidation,25 or depletion of reduced glutathione (GSH), and subsequent increases in cytosolic oxidized glutathione (GSSG) in response to oxidative stress rapidly ubiquitinate NF-κB, which is a critical step for its activation.26
INHIBITORY κB (IκB)
IκB has several isoforms (IκB-α, IκB-β, IκB-γ, IκB-δ, IκB-ε), the most predominant being the α and β subunits.10 These inhibitors have 5–7 conserved domains known as ankyrin repeats, each consisting of around 30 amino acids, forming a unit which is able to interact with NF-κB subunits, thus masking the nuclear localisation signals and preventing activation of NF-κB and its translocation to the nucleus.10 The IκB-α and IκB-ε genes have an κB recognition sequence in their promoter regions.10 ,27 Thus, agents such as phorbol esters, which activate NF-κB, will also induce the synthesis and activation of IκB-α and IkB-ε which enters the nucleus to bind to activated NF-κB and causes the return of NF-κB to the cytoplasm, thereby terminating gene activation.28 The importance of the inhibitory effect of IκB is shown by the fact that disruption of IκB-α in mice results in prolonged activation of NF-κB in response to inflammatory stimuli. In addition, animals treated in this way die of widespread inflammation.29
The IκB family contains other members, including Bcl-3, IκB-γ, and IκB-δ.30 The relative importance of these different inhibitors in regulating NF-κB activation is not clear, but the presence of multiple inhibitors is likely to be important in balancing the NF-κB activation and specific gene regulation.
INHIBITORS OF NF-κB
Several inhibitors of NF-κB activation have been shown to function by either preventing IκB degradation or by inducing IκB-α synthesis (table 2). The immunosuppressive and anti-inflammatory actions of glucocorticoids have been shown to be mediated by the induction of IκB-α synthesis.31 A variety of other compounds can also inhibit NF-κB activation including tepoxalin, a dual inhibitor of cyclo-oxygenase and 5-lipoxygenase,32 the anti-inflammatory cytokine interleukin 10 (IL-10),33 and gliotoxin derived fromAspergillus fumigatus,34However, the mechanism and specificity of inhibition of NF-κB by these agents remains unknown.
Inhibitors of NF-κB
Antioxidants, such as the physiological intracellular redox regulators glutathione and thioredoxin, thioredoxin peroxidase, and the radical scavengers pyrrolidine dithiocarbamate (PDTC) and N-acetyl-l-cysteine (NAC), inhibit NF-κB activation by preventing the inducible decay of IκB-α in response to various stimuli.35 ,36
IκB KINASE(S)
During the process of activation of NF-κB several signal transduction pathways are involved (fig 2). Recent studies have revealed the complexity of this process by demonstrating direct or indirect involvement of a number of known kinases in the phosphorylation event(s), particularly NF-κB inducing kinase (NIK) which has serine/threonine kinase activity homologous to the mitogen activated protein-3-kinase (MAP3K) related kinase.37However, a number of other kinases are possibly also involved in the activation of NF-κB DNA binding activity and κB site dependent transcription. All of the stimuli which activate NF-κB act by means of kinases that phosphorylate and thus degrade IκB by a proteasome mediated mechanism.11 Recently, a serine/threonine kinase, CHUK, downstream of NIK, which is an NIK activated IκB-α kinase, has been characterised.38 Similarly, a cytokine responsive IκB kinase (IKK), almost identical to CHUK, has been described which specifically phosphorylates the critical serines 32 and 36 present on the IκB-α molecule, resulting in activation of NF-κB in response to proinflammatory cytokines.39 The signalling pathways that lead to activation of NF-κB are even more complex because of the large number of different signals involved in its activation. It is possible that multiple signalling pathways become integrated to act on a novel multiprotein IκB kinase complex. In the light of this, a large active multiprotein complex—the IκB kinase (IKK)—consisting of IKK-1 (identical to CHUK) and IKK-2 has been identified, which contains cytokine inducible IκB kinase activity that phosphorylates both IκB-α and IκB-β to form the active heterodimers.40 ,41 However, intriguing questions on the composition, regulation, and function of the IκB kinase complex in response to diverse stimuli remain to be answered—for example, this complex may not be the unique integrator of the NF-κB response so that certain stimuli such as oxidants may follow other pathways to IκB phosphorylation and NF-κB activation.
{kind=link}
{kind=link}
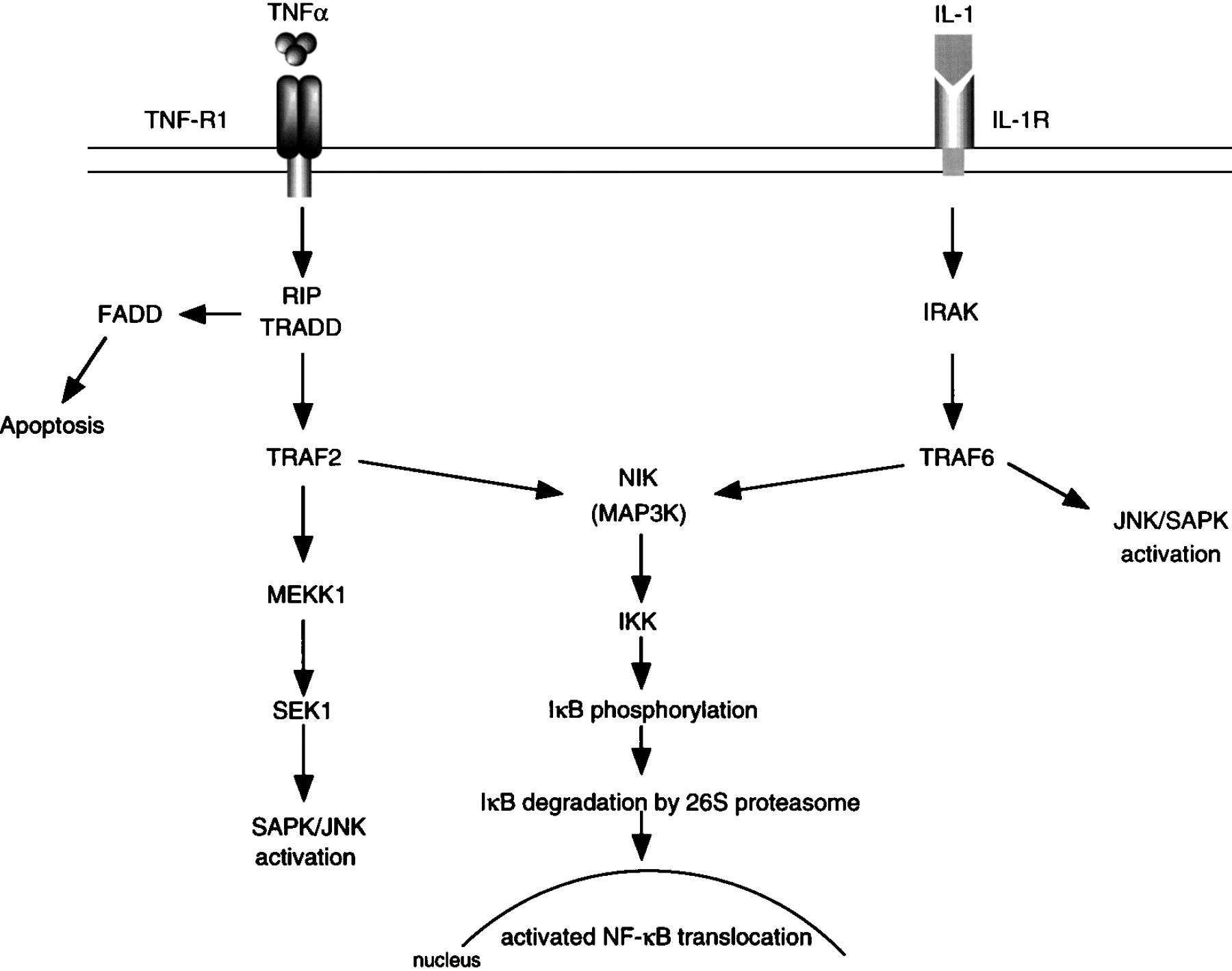
Model for the putative pathways for the activation of NF-κB initiated by TNF-α and IL-1.
REGULATION OF NF-κB BY TNF AND IL-1
It is recognised that tumour necrosis factor α (TNF-α) and IL-1β are pro-inflammatory cytokines involved in the pathogenesis of various inflammatory diseases. TNF-α and IL-1β induce loss of IκB from the cytoplasm, which is preceded by IκB phosphorylation and polyubiquitination of both p105 and IκB-α.42 TNF-α and IL-1β activate NF-κB via distinct families of cell surface receptors and protein-protein interactions (fig 2).38However, both pathways utilise members of the TNF receptor associated factor (TRAF) family of adaptor proteins as signal transducers, and both pathways converge at the level of the protein kinase NIK.37 TNF dependent trimerisation of the TNF receptor leads to recruitment to the cell membrane of adaptor protein which is a TNF-R associated death domain protein (TRADD), receptor interacting protein (RIP) which is a death domain containing serine/threonine kinase and, subsequently, TRAF2, and NIK. This sequence of events results in activation of NIK, which in turn activates the IκB kinase, coupled through phosphorylation of IKK-α and IKK-β in the MAPKK activation loop. This activation may occur at the cell membrane or after active NIK is released from the receptor complex. IκB-α is then recruited to the activated IκB kinase complex where it is phosphorylated by the IKK-α/IKK-β heterodimer. The phosphorylation of IκB-α leads to its ubiquitination and degradation by the proteasome, culminating in the translocation of active NF-κB into the nucleus. In the case of the IL-1 receptor, an interaction between TRAF6 and NIK leads to the activation of the kinase complex. An alternative signal pathway that involves a mitogen activated protein kinase/ERK kinase 1 (MEKK1) or MAPKKK is also possible because MEKK1 is recruited to the TNF-α activated IκB complex.43
TRANSIENT VERSUS PERSISTENT NF-κB ACTIVATION
Although IκB-β interacts as well with the Rel protein dimers as IκB-α, the two proteins display distinct responses to different NF-κB inducers. For example, TNF-α or phorbol myristate acetate (PMA) cause rapid activation of NF-κB and transient and rapid degradation of IκB-α, but not of IκB-β.44 The transient response is due to the fact that activation of NF-κB by these stimuli upregulates the expression of IκB-α. Thus, newly synthesised IκB-α can feedback inhibit NF-κB. By contrast, induction of NF-κB by IL-1β or LPS results in degradation of both IκB-α and IκB-β, and the activity of NF-κB persists longer following stimulation despite the presence of newly synthesised IκB-α and IκB-β. This is because IκB-β is resynthesised as a hypophosphorylated form with an altered conformation that leaves the nuclear localising signal on NF-κB exposed.45 The newly synthesised IκB-β therefore facilitates transport of NF-κB as a stable complex to the nucleus for DNA binding in a manner that protects it from cytosolic IκB-α. Thus, the continued expression of NF-κB as a result of the hypophosphorylated form of IκB-β may lead to persistent expression of the genes induced by IL-1 and LPS.44 The mechanism which prevents newly synthesised IκB-α associating with activated NF-κB in the cytosol to inhibit its translocation remains to be elucidated. It may be that the rate of degradation of the two newly synthetised IκBs are different, such that IκB-α is degraded faster as a more efficient substrate for the IKK-α kinase subunit compared with IκB-β.11 Recent studies in the Drosophila homologue of the mammalian IκB-α, a phosphoprotein cactus, show that it is rapidly phosphorylated and degraded subsequent to its biosynthesis in response to signaling.46 A further explanation may be that in immune responses, when interferon-gamma (IFN-γ) is present, it may not activate NF-κB itself but may synergistically enhance TNF-α induced persistent NF-κB activation by a mechanism that involves increased IκB-α degradation and de novo degradation of IκB-β.47
ROLE OF NF-κB IN PRO-INFLAMMATORY PROCESSES
NF-κB regulates the expression of many genes involved in inflammatory responses in the lungs (table3).5 ,17 ,48 ,49 In all inflammatory diseases adhesion molecules have a role in recruiting inflammatory cells such as neutrophils, eosinophils, and T lymphocytes from the circulation to the site of inflammation.50 NF-κB regulates the expression of several genes that encode adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin. Cytokine induced cell surface expression of E-selectin, VCAM-1 and ICAM-1 and the enhanced secretion of IL-8, monocyte chemoattractant protein 1 (MCP-1), MHC proteins, and other chemokines are regulated at the transcriptional level in endothelial cells by the binding of NF-κB to its putative site in the 5′ flanking sequences.16
Inflammatory genes regulated by NF-κB
In many inflammatory lung diseases such as chronic bronchitis, IPF, ARDS, and human immunodeficiency virus (HIV) depletion of intracellular GSH or increased levels of GSSG are present concomitant with the induction of inflammatory mediators and chemotactic cytokines.51 ,52 This suggests that the intracellular redox state of the cell may have a key role in the regulation of NF-κB and the consequent potentiation of the inflammatory immune responses in lung cells. As has been mentioned earlier, NF-κB is involved in the regulation of genes such as E-selectin, VCAM-1, ICAM-1, IL-8, IL-6, and GM-CSF. These molecules are downregulated in endothelial cells by treatment with antioxidants.53 This is supported by studies of animal models of endotoxin induced NF-κB activation in which administration of the antioxidant NAC downregulates VCAM-1 expression and neutrophil mediated inflammation in the lungs.54 This suggests a central role for NF-κB in the activation of genes whose products promote the adhesion and extravasation of leucocytes at sites of inflammation. Hypoxia is also known to alter intracellular redox state leading to the induction of NF-κB and AP-1 DNA binding,55 which is associated with the induction of ICAM-1, iNOS, and IL-8 gene expression in endothelial cells and monocytes.56-58 Redox regulation of NF-κB may therefore have a pivotal role in cells of the immune system by acting as a potent and pleiotropic transcriptional activator.
ROLE OF NF-κB IN APOPTOSIS
Apoptosis is an important process in the resolution of inflammation. A role of NF-κB in this process has also been suggested. TNF-α, which is a strong inducer of NF-κB, also triggers apoptosis via TNF receptor 1.59 However, the pathways leading to apoptosis and NF-κB activation may be distinct. By contrast, activation of NF-κB has been shown to suppress TNF-α mediated apoptosis.60 These findings suggest that pro-apoptotic extracellular signals can also induce NF-κB, which in turn probably induces expression of genes that are anti-apoptotic. It appears that the role of NF-κB in the regulation of apoptosis differs from cell to cell depending on their signalling pathways.61
ACTIVATION OF NF-κB IN INFLAMMATORY LUNG DISEASES
There is clear evidence for the upregulation of the genes for pro-inflammatory mediators in a number of inflammatory lung diseases. Activation of NF-κB has been shown in human peripheral blood mononuclear cells, airway epithelial cells (1HAEω-) and lung tissue in response to proinflammatory cytokines such as IL-1β, TNF-α, or in response to oxidants.62 ,63 However, there have been relatively few direct measurements of the activation of NF-κB in inflamed lungs. NF-κB is activated in submucosal cells, in sputum derived macrophages, in endothelium, and in epithelial cells in bronchial biopsy specimens from patients with asthma.64 It has been reported that in alveolar macrophages from patients with ARDS NF-κB is more activated than in alveolar macrophages from critically ill patients with other diseases.65 However, experimental evidence linking NF-κB activation to the human lung inflammatory diseases is lacking.
Several animal models have been developed to evaluate the role of NF-κB in lung inflammation. Intraperitoneal administration of endotoxin in rats leads to the activation of NF-κB in alveolar macrophages and in lung tissue which is associated with lung neutrophilia, epithelial permeability, and lipid peroxidation.54 In a mouse model shock caused by haemorrhage produces rapid activation of the NF-κB and the cyclic AMP response element binding protein (CREB) through xanthine oxidase dependent mechanisms in pulmonary mononuclear cells.66 ,67It has been suggested that activation of NF-κB occurs through α-adrenergic stimulation by a mechanism involving the generation of reactive oxygen species (ROS). This may be the mechanism by which blood loss produces increased cytokine expression in the lungs.68
Activator protein 1 (AP-1)
C-FOS AND C-JUN PROTEINS AND THEIR INTERACTIONS
Activator protein 1 (AP-1) is an important transcription factor composed of proto-oncogenes as heterodimers of c-fos, c-jun or as c-jun/c-jun homodimers or activating transcription factors (ATF). AP-1 is involved in the transcriptional control of many inflammatory mediators. Activated AP-1 has been shown to disrupt the structure of the nucleosome completely.69 Nucleosome disruption is considered to be the important first step in the chromatin remodelling process involved in the initial binding of transcriptional factors to a nucleosomal template which allows the transcriptional machinery to function. Dimerisation of fos-jun or jun-jun is a prerequisite for DNA binding via a “leucine zipper” domain which serves as a crucial regulator of the expression of a wide variety of genes.70However, such interaction is limited to the heterodimers of c-fos and c-jun only. Other members of the fos and jun family of proteins are also known to interact with each other but are unable to activate AP-1 mediated transcription. All of the Jun family of proteins are capable of forming homodimers and heterodimers which can bind to a cognate DNA site. Fos proteins do not associate with each other, but are capable of associating with any members of the Jun family to form stable dimers that have higher DNA binding activity than Jun dimers.
REGULATION OF AP-1
Activation of the tumour promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) response element (TRE; this element has the base sequence TGACTCA) is a prerequisite for AP-1 induced transcriptional regulation. Hence, any agents which activate TRE, such as serum responsive factor, also activate AP-1 by a protein kinase C mediated pathway. Evidence suggests direct involvement of ROS in AP-1 activation by the c-jun N-terminal kinases (JNKs), which are members of the MAP kinase superfamily.71 TNF-α induced ROS production acts as a common signal to stimulate AP-1 activation and gene expression by a novel stress activated protein kinase (SAPK)/JNK.72 Mitogen induced AP-1 activation has been shown to be antioxidant inhibitable, suggesting that it is ROS mediated.73
However, little is known of the exact mechanism underlying ROS dependent AP-1 activation. Perturbation of cellular thiol redox status has been suggested as a signal that may be involved in the induction of c-fos and c-jun expression, since the intracellular GSH/GSSG ratio is a key regulator for the induction of stress activated signal transduction pathways by JNK and p38 kinase.74 Depletion of intracellular GSH in airway epithelial cells following exposure to cigarette smoke free radicals leads to increased AP-1 DNA binding75 and release of IL-8.76 Inhibition of AP-1 activation by the antioxidant NAC, as well as prevention of the induction of c-fos and c-jun mRNA, has also been shown to occur in mesothelial cells following treatment with NAC, suggesting a direct interaction of oxidants and antioxidants with specific cysteine groups which are required for AP-1 binding. However, contradictory data have been obtained concerning the mode of AP-1 regulation by reduced thiols, such that NAC enhanced AP-1 DNA binding affinity and transactivation.35 Certain phenolic antioxidants such as butylated hydroxytoluene and butylated hydroxyanisole exhibit anti-inflammatory effects and have been observed to increase the expression of the c-fos and c-jun mRNA substantially and to induce AP-1 DNA binding.77 Clearly the relationship between oxidants/antioxidants and transcription factor regulation is complex and requires further study.
ROLE OF AP-1 IN INFLAMMATORY PROCESSES
AP-1 activation is essential for cellular proliferation and differentiation.70 However, data suggesting a role for AP-1 in inflammatory responses are few. Recent data indicating that redox sensitive JNK and p38 are activated by pro-inflammatory cytokines, such as TNF-α and IL-1, suggest that these signalling pathways may also have a role in inflammatory responses. Pharmacological inhibitors of p38 block many inflammatory responses, especially the production of cytokines IL-1, TNF-α, and IL-6.78 The promoters of many inflammatory response genes, especially those encoding cytokines and chemokines, have AP-1 binding sites. This suggests a possible role for JNK in their regulation. JNK is also thought to be involved in the induction of cyclo-oxygenase 2/prostaglandin synthase 2 which plays an important part in the inflammatory response by catalysing the production of prostaglandins.79 Platelet activating factor (PAF) has been implicated in the pathogenesis of allergic and inflammatory events in the airways. Binding of PAF to its receptor on human bronchial epithelial cells leads to activation of AP-1-mediated transcription.80
Direct measurements of components of AP-1 in disease processes are lacking. AP-1 and its components c-fos and c-jun are overexpressed in lungs of smokers.81 Expression of c-jun and c-fos were induced in alveolar macrophages and type II pneumocytes in a rat model of lung fibrosis after intratracheal administration of bleomycin.82 Interestingly, c-fos is induced in epithelial cells in asthmatic subjects, suggesting a role for AP-1 in the regulation of the inflammation in asthma.83 More work will be needed to determine whether and how the JNK and p38 pathways can mediate inflammatory responses and how the components of AP-1 are involved in the pathogenesis of lung diseases.
Nuclear factor-interleukin 6 (NF-IL6)
NF-IL6 was originally identified as a DNA binding protein responsible for IL-1 stimulated IL-6 induction.84 It has a sequence ACATTGACAATCT and direct cloning of NF-IL6 has revealed that it recognises the same nucleotide sequences as CCAAT enhancer binding protein (C/EBP).7 NF-IL6 is rapidly phosphorylated and synergistically induced by LPS or inflammatory cytokines such as IL-1, TNF, and IL-6. NF-IL6 can also bind to the regulatory region of various genes including IL-8, G-CSF, IL-1, and immunoglobulin genes.85 Furthermore, NF-IL6 has been shown to be identical to IL-6DBP, a DNA binding protein responsible for IL-6 mediated induction of acute phase proteins, demonstrating that NF-IL6 is involved in the expression of the genes regulated by IL-6.7 Like NF-κB, NF-IL6 is also considered to be a pleiotropic mediator of many inducible genes involved in acute, immune, and inflammatory responses, such as the genes for IL-7, IL-8, ICAM-1, iNOS, cyclo-oxygenase-2 genes, and several acute phase protein genes.7
ROLE OF NF-IL6 IN INFLAMMATORY PROCESSES
In normal lung tissue NF-IL6 mRNA and NF-IL6 protein are almost undetectable.86 However, infection of human type II pulmonary alveolar epithelial cells with respiratory syncytial virus (RSV) leads to NF-IL6 gene expression and its synthesis.86NF-IL6 is predominantly expressed in macrophages and is activated in response to IL-1, TNF-α, and LPS. NF-IL6 regulates the E1A of adenovirus responsive promoters.87 Adenovirus E1A proteins modulate ICAM-1 induction by LPS in airway epithelial cells and may amplify the inflammatory processes in the airways of smokers and patients with COPD.88 IL-6 levels have been shown to be higher in bronchoalveolar lavage fluid from patients with lung fibrosis and those with work related histories of long term asbestos exposure.89 In vitro studies on the effect of asbestos fibres on an airway epithelial cell line showed induction of IL-6, which is associated with the activation of both NF-IL6 and NF-κB, which may result from an oxidant induced mechanism.89
Nuclear factor of activated T cells (NFAT)
The nuclear factor of activated T cells (NFAT) was first identified in the IL-2 promoter in human T cells. Transcription factors of the NFAT family regulate the production of effector proteins that coordinate the immune response. Despite their name, NFAT proteins are expressed not only in T cells, but also in other classes of immune system cells ranging from mast cells, monocytes, macrophages, and eosinophils to endothelial cells.6 NFAT proteins are activated by stimulation of receptors coupled to calcium mobilisation. Receptor stimulation and calcium mobilisation result in activation of many intracellular enzymes, including the calcium and calmodulin dependent phosphatase calcineurin, a major upstream regulator of NFAT proteins. Stimuli that elicit calcium mobilisation result in the rapid dephosphorylation of NFAT proteins and their translocation to the nucleus where they have strong binding affinity to DNA. The amplitude and duration of calcium signals in B cells has been shown to control the differential action of NF-κB and NFAT. NF-κB and JNK are selectively activated by a large transient rise in intracellular calcium whereas NFAT is activated by a low sustained rise in calcium.90
ROLE OF NFAT IN IMMUNE INFLAMMATORY PROCESSES
Stimulated T cells transcribe a large array of activation associated genes, many of which are potential targets for NFAT; these genes encode transcription factors, signalling proteins, cytokines IL-2, IL-4, IL-5, IL-8, GM-CSF, TNF-α, cell surface receptors, and other effector proteins.6 NFAT plays an important role in regulating the T helper 1 and 2 (Th1 and Th2) patterns of cytokine production. Th1 cells produce IL-2 and IFN-γ whereas Th2 cells produce IL-4, IL-5, IL-1, and IL-13. Thus, NFAT influences T lymphocyte activation and the cytokine profiles of T cells by differentiating Th1 and Th2 pathways, which may be important in the pathogenesis of many immune disorders. Putative GATA-3, together with NFAT sites, are present on the promoter of IL-5 which may be responsible for the induction of Th2 type cytokines.91 Since IL-5 is produced by CD4+ Th 2 cells, but not by Th1 cells, NFAT may play a key role in the development of eosinophilia in asthma. NFAT proteins show a characteristic ability to interact with AP-1 and NF-κB DNA binding and transactivation. It has been shown that coupled NFAT:AP-1 is more stable and has higher affinity for DNA.92 Such interactions are important for the regulation of IL-2, IL-4, IL-5, CD40L, GM-CSF gene expression.6
As substrates for calcineurin, NFAT proteins are major targets of the immunosuppressive drugs cyclosporin A and FK506 because of their ability to inhibit dephosphorylation of NFAT.93 An understanding of the regulation and function of NFAT proteins may provide us with selective inhibitors of individual NFAT proteins in specific cell types or for selected inducible genes as a means of modulating immune responses.
Intracellular interactions of transcription factors involved in pro-inflammatory processes
The expression of most genes is controlled by the concerted action of multiple distinct transcription factors. For instance, the promoter of the IL-2 gene contains DNA binding sites for at least four distinct transcription factors: NF-κB, AP-1, NFAT, and Oct factors.94 Some of these factors can physically interact with each other when bound to DNA and thereby synergistically promote DNA binding and transactivation. For example, NF-κB has been shown to interact directly with AP-1 subunits95 and with NFAT within the IFN-γ promoter,96 whereas AP-1 coordinately interacts with the ets transcription factor in the collagenase promoter.97 NFAT also binds AP-1 and recruits this factor to DNA.6 NF-κB is also known to synergise with a number of different transcriptional activator proteins, including Sp1, ets, c-Jun, and NF-IL6. This synergy is a consequence of the direct interactions between NF-κB and these proteins and cooperative binding to adjacent binding sites. The interactive transcription factor can be present constitutively or co-induced with NF-κB. An example of the latter is the recent finding that several inducers of NF-κB activate the c-Jun N-terminal protein kinase (JNK)/p38 which, in turn, phosphorylates c-Jun and synergises with NF-κB.98Upregulation of E-selectin gene expression by TNF-α and induction of IL-8 and iNOS gene may also involve a similar mechanism.99 ,100 On the other hand IL-4, which is an immunoregulatory cytokine secreted from activated Th2 lymphocytes, eosinophils, and mast cells, activates the transcription factor STAT6101 which competes for binding to NF-κB binding sites. This competition leads to suppression of TNF-α induced E-selectin gene expression in human vascular endothelial cells.101 Thus, physical interactions between transcription factors modulate the expression of numerous inflammatory products such as adhesion molecules, cytokines, chemokines and growth factors, which are involved in the inflammatory responses in lungs.
Anti-inflammatory effect of glucocorticoids on transcription factors
Glucocorticoids have been used for decades to suppress both the immune response and inflammation, yet the immunosuppressive mechanism by which these drugs act is poorly understood. There is increasing evidence that glucocorticoids inhibit the action of transcription factors such as AP-1 and NF-κB.102 Glucocorticoids increase the transcription of the gene encoding IκB-α and thus suppress the inflammatory process by inhibition of IL-2, IL-6, IL-8, E-selectin, and iNOS gene expression.103 ,104 In the cytoplasm glucocorticoids activate an intracellular receptor protein, the glucocorticoid receptor. Binding of the glucocorticoid to its receptor transduces the hormone signal to the nucleus as a transcription factor.102 Here it binds as a homodimer to glucocorticoid response elements (GRE) in steroid responsive target genes, resulting in either induction or repression of gene transcription. However, glucocorticoids decrease the transcription of the genes involved in inflammation which have no identifiable glucocorticoid response elements in their promoter regions, suggesting that some other mechanism must mediate some of the inhibitory effects of these hormones. One explanation is that there may be a direct protein-protein coupling/interaction between the glucocorticoid receptor and AP-1105 and between the receptor and NF-κB.106 Thus, the activated glucocorticoid receptors may then bind to activated NF-κB, AP-1, or CREB and prevent these transcription factors from binding to their respective consensus regions on genes. These interactions may occur in the cytoplasm or the nucleus.107 Thus, glucocorticoids are potent inhibitors of the activation of NF-κB and AP-1, which may account for most of their anti-inflammatory actions. However, the relative importance of these different mechanisms to the immunosuppressive/anti-inflammatory actions of glucocorticoids remains to be established. Some in vivo support for this hypothesis comes from studies in mild asthmatic subjects in whom treatment with corticosteroids suppressed the activation of NF-κB and components of AP-1 in the bronchial mucosa obtained by bronchial biopsy.83
Since glucocorticoids inhibit both NF-κB and AP-1 activities, although by different mechanisms, it is not surprising that they decrease expression of a very wide range of immunoregulatory genes. It is also likely that they regulate apoptosis by direct physical interactions with p65/AP-1. However, glucocorticoids have opposite effects on apoptosis in neutrophils and eosinophils.108
Nuclear receptor co-activators
Steroid receptors and co-activator proteins are known to stimulate gene expression by facilitating the assembly of basal transcription factors into a stable pre-initiation complex. Thus, the transcription factors gain access to transcriptionally repressed chromatin to modulate the transactivation of specific gene networks. Acetylation of chromatin in vivo is coupled to transcription. Specific histone acetyltransferases (HATs) target histones bound to DNA and overcome the inhibitory effect of chromatin on gene expression. High levels of histone acetylation and linker histone deficiency are correlated with gene activity and reduced levels with gene silencing.109 A few proteins have been identified as nuclear HATs; these include steroid receptor co-activator 1,110 p300/CBP associated factor (P/CAF),111 p300/CREB-binding protein (CBP),112 and TAFII230/250.113
CREB-BINDING PROTEIN (CBP ) AND P300
CBP and adenovirus E1A associated protein p300 play essential co-activator roles for a number of transcription factors including CREB, AP-1, GR, and STAT.8 CBP and p300 could interact simultaneously with more than one class of transcription factor. The amounts of these co-activator proteins are tightly controlled. Thus, for promoters that are regulated by CBP dependent transcription factors, rates of transcription will depend not only on the levels and activation state of bound transcription factors, but also on the cellular levels of CBP/p300 and the extent to which these co-activators are utilised elsewhere. For example, the glucocorticoid receptor is the most efficient inhibitor of AP-1 activity.105
Recent studies have suggested that CBP and p300 also function as essential co-activators for several nuclear receptors including glucocorticoid receptors.110 Hence, glucocorticoid receptor antagonism of AP-1 activation may occur by competition of glucocorticoid receptors with these co-activators and therefore accounts for transrepression of genes.114
STEROID RECEPTOR CO-ACTIVATOR 1 (SRC-1)
SRC-1 is a co-activator for many members of the steroid hormone receptor superfamily of ligand inducible transcription factors. SRC-1 possesses intrinsic HAT activity. It also interacts with another HAT, p300/CBP associated factor (P/CAF).110
It is likely that these co-activators and P/CAF are recruited to the promoter regions of steroid responsive genes in vivo by interaction with steroid receptors that bind specific DNA response elements in response to ligand. The basal levels of transcription of most genes appear to be maintained by histone deacetylation which limits access to transcription factors and represses gene activation.115However, intrinsic acetyltransferase activity of co-activators such as SRC-1, CBP and P/CAF may alter the existing equilibrium between histone acetylation and histone deacetylation towards the progressive accumulation of nucleosomes containing acetylated histones.115 This targeted histone acetylation may contribute directly to the transcriptional activation process by disrupting the repressive chromatin structure and allowing formation of the pre-initiation complex in the region of the promoter nucleosome which contains the TATA box and/or initiator element. It is, however, unclear how histone acetylation and deacetylation in vivo affect steroid receptor transduction of gene expression.
Implications of glucocorticoid receptor binding with nuclear co-activators in inflammatory processes
In many chronic inflammatory disorders, such as asthma, glucocorticoid insensitivity is a rare but challenging clinical problem. The molecular basis of glucocorticoid insensitivity, however, is unknown. Abnormal numbers and availability or recruitment of nuclear receptor co-activators, their antagonism with other basal transcription factors such as AP-1, or their intrinsic acetyltransferase/deacetylase activity and chromatin remodelling, may have an important role in controlling the rate of transcription/repression of genes in various inflammatory lung diseases. In patients whose asthma is resistant to the anti-inflammatory effects of glucocorticoids, there appears to be exaggerated activation of AP-1. NF-κB and β2 agonists activate CREB that binds to and therefore sequesters activated glucocorticoid receptors inside the nucleus.116 This will reduce the availability of glucocorticoid receptors to inhibit NF-κB or AP-1 or CREB which are normally active in such patients. It is also possible that these abnormal glucocorticoid receptors will not repress the transcription machinery due to alteration in HAT activity of co-activators which have impaired binding to the glucocorticoid response element. The precise role of glucocorticoids on various transcription factors and co-activators in the regulation of various cytokines, particularly in the inflammatory process, is an intense area of investigation. Understanding the mechanisms of the transcriptional machinery in normal and inflammatory disease states should provide a molecular mechanism for a novel approach to therapy.
Therapeutic implications
As described above, NF-κB and AP-1 are activated by many of the factors which enhance the inflammatory response. This activation in turn leads to the coordinated expression of many genes that encode proteins involved in mediator synthesis and the further amplification and perpetuation of the inflammatory response. NF-κB and AP-1 are therefore obvious targets for novel anti-inflammatory treatments. Glucocorticoids are effective inhibitors of NF-κB and AP-1 but have endocrine and metabolic side effects when given systemically. More specific NF-κB and AP-1 inhibitors could be developed which may have less serious side effects. Currently there is much interest in identifying more specific and effective NF-κB inhibitors.20 These include specific targets in the signalling pathway such as cytokine receptors, the DNA-binding NF-κB dimer itself, ubiquitin conjugating enzymes, and the proteasome. The most obvious and specific choice would be a cytokine responsive IκB multiprotein kinase complex (IKK)-IκB-α and IκB-β kinases which have been recently shown to activate the IκB in response to TNF and IL-1.117 It is probable that screening for specific IKK inhibitors, in particular, which modulate the activation and function of the IKK (IKK-1/2) is likely to have therapeutic value in the modulation of inflammatory responses. In addition, identification of the other components of the IκB kinase complex will allow this to be targeted with novel drugs. It may also be possible to target various IκBs which regulate subsets of NF-κB responsive genes. A better understanding of JNK and p38 pathways in the mechanism of activation of AP-1 is required for the development of more specific anti-inflammatory drugs against the genes regulated by AP-1.
Biochemical and clinical studies indicate that antioxidant therapy may be useful in the treatment of a wide variety of diseases by optimising the redox and antioxidant defences, or by inhibiting NF-κB activation.118 Activation and binding of transcription factors to consensus sites on DNA may be driven by the physiological redox homeostasis, especially by the intracellular thiol-disulphide balance. The endogenous glutathione and thioredoxin systems may therefore be target molecules to modulate the expression of redox sensitive cytokine, interleukin, and adhesion molecule gene expression.118
It may not, however, be logical to block the activation of NF-κB for prolonged periods since it plays such a critical part in the immune and other defence responses. Targeted disruption (or knockout) of the p65 component of NF-κB is lethal in animal models because of associated developmental abnormalities119whereas the lack of the p50 component results in immune deficiency and increased susceptibility to infection.13 Since NF-κB often works in concert with other transcription factors, it may be possible to achieve a more selective blockade in specific cell types in a particular disease or a blockade of a restricted set of genes by developing compounds that inhibit the synergistic interactions between several transcription factors. For example, although there are many similarities between the inflammatory responses in patients with asthma and other inflammatory lung diseases such as COPD, there are also important differences in the type of inflammatory cells involved and in the mediators of the inflammation. These differences may relate to the secretion of specific cytokines such as IL-5, which promotes eosinophilic inflammation in patients with asthma, and TNF-α and IL-8 which cause neutrophil influx in lungs of patients with COPD. Further research on the interactions between transcription factors which might mediate these differences in inflammatory lung diseases is needed. Further studies are also required to determine whether the intensity of NF-κB activation is useful as a marker for the severity of lung inflammation. It is hoped that the investigation of compounds capable of inhibiting the activation of transcription factors may lead to the development of novel treatment strategies targeted at the specific inflammatory responses in chronic inflammatory lung diseases.
Acknowledgments
Supported by the British Lung Foundation. The authors wish to thank Mr Mark Lawson and Miss Tammy Watchorn for drawing the original figures.