Article Text

Abstract
Langerhans’ cell histiocytosis may cause irreversible respiratory failure due to progressive destruction of lung parenchyma and widespread cystic change. Transplantation offers a therapeutic option. A case is described of recurrence of Langerhans’ cell histiocytosis which was associated with deterioration in lung function four years following bilateral lung transplantation. Patients transplanted for Langerhans’ cell histiocytosis should be followed up with this complication in mind.
Statistics from Altmetric.com
Case report
An otherwise fit 27 year old Asian male martial arts instructor presented in mid 1991 with recurrent spontaneous pneumothorax. He smoked 10 cigarettes per day and was taking no medication. A diagnosis of pulmonary Langerhans’ cell histiocytosis was made based on the clinical picture and on the radiological findings. Chest radiography and high resolution computed tomographic scanning showed reticular shadowing and widespread cystic changes in the lower lobes. A lung biopsy specimen showed reactive pleuritis and interstitial emphysema but no evidence of active or burnt out Langerhans’ cell histiocytosis. The skull radiograph was normal and a bone scan showed non-specific changes.
Lung function deteriorated over the next two years, with reduction in forced expiratory volume in one second (FEV1) to 0.88 litres (22% predicted) and of forced vital capacity (FVC) to 2.04 litres (44% predicted). In April 1993 he underwent double lung transplantation with internal mammary artery revascularisation. The diagnosis of Langerhans’ cell histiocytosis was confirmed on histological examination of the explanted lungs, showing florid active disease as well as cystic change consistent with burnt out Langerhans’ cell histiocytosis. The active lesions consisted of collections of non-pigmented mitotically active Langerhans’ cells interspersed with eosinophils and lymphocytes.
Postoperative recovery was complicated by pneumonia due toPseudomonas aeruginosa and cytomegalovirus pneumonitis, both conditions responding to appropriate therapy. Immunosuppression consisted of cyclosporin and azathioprine with initial oral prednisolone which was slowly withdrawn over the first year. At his first annual assessment he was clinically stable. Selected mammary artery angiography showed a patent anastomosis into the bronchial circulation, chest radiography was unremarkable and respiratory function tests were normal, with an FEV1 of 3.31 litres (87% predicted) and an FVC of 3.91 litres (88% predicted). He had stopped smoking after his transplant.
However, 11 months later his respiratory function had deteriorated, with an FEV1 of 2.72 litres (72% predicted) and an FVC of 3.63 litres (79% predicted). Bronchoscopic examination showed normal airways, the bronchoalveolar lavage fluid was normal, and the transbronchial biopsy specimen—which included bronchiolar wall as well as lung parenchyma—showed no evidence of rejection, infection, or obliterative bronchiolitis. Lung function failed to improve after a three day course of intravenous methylprednisolone and subsequent oral prednisolone. A technetium-99 diethylene triamine penta-acetic acid (99Tc-DPTA) lung ventilation scan showed an area of abnormal accumulation of contrast in the right lung base. This corresponded with ill defined shadowing seen on the chest radiograph. There was no central deposition of the 99Tc-DPTA in the major airways. High resolution computed tomographic scanning of the lungs revealed widespread cystic changes in both lung fields involving the mid and lower zones suggestive of recurrent Langerhans’ cell histiocytosis (fig 1). This was confirmed on a transbronchial biopsy specimen from the right lower lobe. There were interstitial, peribronchiolar and perivascular clusters of S100 positive Langerhans’ cells with eosinophils and lymphocytes, similar to the findings in the explanted lungs (fig 2). He did not tolerate a course of chemotherapy (doxorubicin, vincristine and etoposide), given in an attempt to arrest disease activity. His lung function deteriorated despite treatment with 15 mg prednisolone in addition to cyclosporin and cyclophosphamide but is now stable. Four years after transplantation his FEV1 is 2.19 litres (51% of predicted) and his FVC is 3.53 litres (77% predicted).

Representative high resolution computed tomographic views of the left base of (A) the recipient lung and (B) the donor lung 23 months after double lung transplantation. Both views show multiple parenchymal cystic spaces.

{kind=link}
{kind=link}
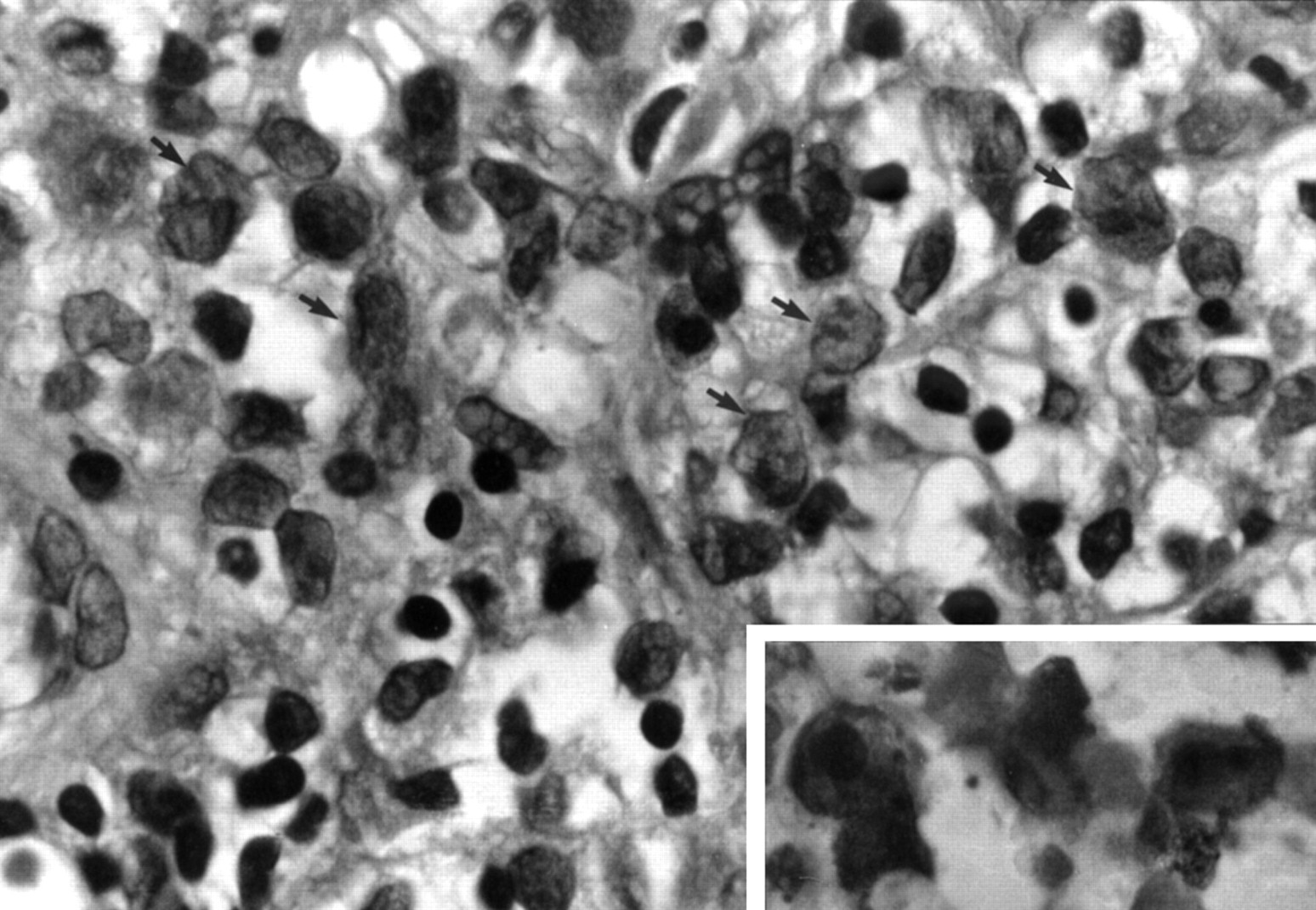
Transbronchial lung biopsy specimen taken 23 months after double lung transplantation showing interstitial aggregates of Langerhans’ cells with large nuclei having fine chromatin and delicate folds and grooves (arrows), and pale-staining cytoplasm with indistinct cell borders. Stain: haematoxylin and eosin. Original magnification ×400. Inset: Immunostaining for S100 protein shows strong membrane and cytoplasmic positivity of the Langerhans cells. Stain: streptavidin-biotin. Original magnification ×1000.
Discussion
Langerhans’ cell histiocytosis is a heterogeneous group of conditions of unknown aetiology characterised by an abnormal proliferation of antigen presenting cells of bone marrow derivation known as Langerhans’ cells.1 Isolated lung involvement is most commonly seen in young adults, many of whom are cigarette smokers. Light microscopy shows scattered discrete nodules that are frequently bronchiolocentric. The nodules are formed of focal collections of Langerhans’ cells characterised as such by their morphology (nuclei with fine chromatin and grooves or folds), immunophenotype (S100 and CD1 positive) and ultrastructure (cytoplasmic Birbeck granules), interspersed with eosinophils and small lymphocytes.1-3The outcome is highly variable, ranging from rapid spontaneous resolution to irreversible respiratory failure. The infiltrate leads to progressive destruction of lung parenchyma and the development of widespread cystic change.1 ,2 Initially many patients are asymptomatic but progressive dyspnoea may occur as the cysts enlarge and coalesce. There is a high incidence of pneumothorax. As there is no definitive treatment, pulmonary Langerhans’ cell histiocytosis has recently been added to the list of indications for lung transplantation.4 The potential for recurrence of disease in the donor organ has long term implications for graft function and survival.
There are a number of noteworthy features in our case. Firstly, the disease typically affects the upper and middle lobes and spares the costophrenic angle5 ,6 but in our patient the radiographic changes were most prominent in the lower lobes, both initially and on recurrence. Secondly, it is reported that this condition is rare in Asian patients7 but our patient was born in Pakistan of Asian parents and moved to the UK at six years of age. Lastly, respiratory function studies in Langerhans’ cell histiocytosis often shows an obstructive pattern, reflecting its bronchiolocentricity but overlapping with the obliterative bronchiolitis syndrome8which remains the most serious long term complication of lung transplantation.9 A contribution by the bronchiolitis obliterans syndrome to deteriorating lung function in our patient cannot be excluded despite histological confirmation of disease recurrence.
Although the aetiology of pulmonary Langerhans’ cell histiocytosis is unknown, its bronchiolocentricity and tendency to regress following cessation of cigarette smoking suggests a reactive immune response in the bronchioles mediated by the Langerhans’ cell, possibly through cytokine production.10 The suggested antigen is unknown but is presumed to be inhaled. The recurrence of the disease after transplantation suggests either that extrapulmonary factors play a role in pathogenesis or that, in this instance, the disease may be truly neoplastic. The recent demonstration that Langerhans’ cells in pulmonary Langerhans’ cell histiocytosis may proliferate locally, showing an abnormal phenotype, lends some support to the latter theory.11 Why the disease should recur in exactly the same pulmonary distribution before and after transplantation in our patient is unclear.
Recent reports have suggested a possible role for cyclosporin in the treatment of Langerhans’ cell histiocytosis12 as it is known to induce cytokine mediated cellular activation selectively. Although the disease recurred in our patient despite standard immunosuppressive therapy following transplantation, which included cyclosporin, it is possible that cyclosporin slowed down the rate of progression of the disease.
In summary, this report documents recurrent Langerhans’ cell histiocytosis after double lung transplantation. Such recurrence may be associated with clinical deterioration. We suggest that other patients transplanted for this condition be followed up with this complication in mind.