Article Text

Statistics from Altmetric.com
The lung is frequently involved in primary vasculitis especially in Wegener’s granulomatosis. Pulmonary involvement can occasionally occur alone but is more commonly seen as part of a more generalised disease. The eyes, ears, nose, sinuses, oral cavity and salivary glands are other common targets of injury. The trachea and bronchi may be affected with inflammatory pseudotumour leading to subglottic stenoses of the trachea and bronchial stenoses are well recognised. Renal involvement is frequently found and only rarely occurs without evidence of other organ involvement. The skin, joints, central and peripheral nervous system, gastrointestinal tract, genital tract, spleen, and heart may all be targeted in the inflammatory process. Pulmonary involvement per se may give rise to solitary or multiple nodules which often cavitate and can be surrounded by thick walls of inflammatory tissue. Localised or diffuse pulmonary infiltrates, lobar collapse, atelectasis, and alveolar haemorrhage are other common manifestations of pulmonary involvement in systemic vasculitis. Pleural effusions and inflammatory pleural pseudotumours can also occur.
The discovery that antineutrophil cytoplasmic antibodies (ANCA) are strongly associated with vasculitic disorders and their subsequent characterisation has led to improved understanding of the pathogenic mechanisms underlying vasculitis. This paper will review clinical aspects of pulmonary involvement in the systemic vasculitides and explores the impact which the discovery of auto-antibodies in these disorders has had on our understanding of their pathogenesis. It also investigates how these discoveries can be used to improve the management of patients with these conditions.
Antineutrophil cytoplasmic antibodies
Antineutrophil cytoplasmic antibodies (ANCA) are circulating autoantibodies which identify a specific subpopulation of patients with systemic vasculitis. These autoantibodies were originally detected by immunofluorescence and two distinct patterns were described: cytoplasmic or c-ANCA which are directed against proteinase-3, and perinuclear (p-ANCA) which is usually directed against myeloperoxidase although other antigenic targets, the nature and relevance of which will be discussed below, have been described.1-7 The term “atypical ANCA” is also used to describe fluorescent patterns which are neither classical nor perinuclear and are usually found in patients with a variety of inflammatory disorders such as inflammatory bowel disease, primary sclerosing cholangitis and rheumatoid arthritis.1 8 Proteinase-3 and myeloperoxidase usually reside within azurophil granules in the neutrophil and are normally involved in host defence against invading organisms. In reality, therefore “ANCA” now refer to an increasing number of autoantibodies directed against targets within neutrophils and are associated, in general with clinically different, although often overlapping, diseases. Solid phase assays for detection of these antibodies are now widely used and much work has been done to improve their quality and reproducibility.6 9-11
The two main immunofluorescent patterns of ANCA positivity have broadly different disease associations. The c-ANCA is positive in more than 90% of patients with active “classical” Wegener’s granulomatosis. In over 700 normal individuals tested by Hagen et alhardly a single c-ANCA was detected.9-11 In “limited” Wegener’s granulomatosis—that is, without active glomerulonephritis or in “inactive” disease—the sensitivity of the c-ANCA may be as low as 43% or as high as 65–70%.2-5 Although up to 5% of patients with “classical” Wegener’s granulomatosis are found to have p-ANCA, this antibody is usually associated with a number of other vasculitic syndromes including idiopathic necrotising and crescentic glomerulonephritis, Churg-Strauss syndrome, microscopic polyangiitis, and a growing list of other inflammatory disorders.1 8There has been some recent interest in looking for ANCA in bronchoalveolar fluid of patients suspected of having pulmonary vasculitis in whom searches for serum ANCA antibodies have been fruitless.12
The first description of ANCA over 15 years ago and the subsequent association of this phenomenon with Wegener’s granulomatosis has led to an explosion of interest in the scientific and clinical aspects of vasculitis. There is no doubt that ANCA testing has also triggered many controversies, yet now that many of the methodological problems have been sorted out and attempts to standardise ANCA testing are well underway, what has emerged are three important advances: redefinition of disease boundaries, improved understanding of possible pathogenic mechanisms, and improved treatment for patients with these conditions.
Nomenclature
It is certain that circulating ANCA are found in patients with certain forms of necrotising vasculitis and that the titres of these antibodies reflect disease activity, at least in some patients.13-16 However, by 1993 it was abundantly clear that international agreement needed to be reached with regard to nomenclature of the systemic vasculitides if further progress was to be made. An international consensus conference was held in Chapel Hill, North Carolina, USA where the proposal that the vasculitides be divided based on the size of the vessel involved in the inflammatory process emerged.17 Thus, the terms large, medium and small vessel vasculitis emerged. This emphasis on involved vessel size is consistent with the macroscopic and histopathological findings of many of the early workers in this field such as Zeek, Goldman and Churg who tried to separate the necrotising angiitides into clinically and pathologically distinctive categories in the 1940s and 1950s.18-21 Although this broad classification of the vasculitides has been widely accepted, there are some who feel that the childhood vasculitides in particular are not well served by these definitions.
LARGE VESSEL VASCULITIS
The large vessel vasculitides include giant cell (temporal) arteritis and Takayasu arteritis where granulomatous inflammation is confined to the aorta and its major branches, with the predilection for extracranial branches of the carotid artery in the case of temporal arteritis. Temporal arteritis affects mainly those over the age of 50 and is often associated with polymyalgia rheumatica while Takayasu arteritis usually occurs in patients younger than 50. Pulmonary involvement in these forms of vasculitis is not reported although thoracic aortic aneurysms occur in 15% of patients with giant cell arteritis.
MEDIUM VESSEL VASCULITIS
Medium size vessel vasculitis refers to classical polyarteritis nodosa (PAN) and Kawasaki disease. Classic PAN was originally described almost a century and a half ago by the Viennese pathologist Karl Rokitansky who described nodular aneurysms in the arteries of a 23 year old patient.22 Fourteen years later the German partnership of Kussmaul and Maeir—who are often attributed with the first descriptions of PAN—published their elegant description and diagrams of a similar case.23 Medium sized vessels refer to the main visceral arteries—for example, renal, hepatic, coronary and mesenteric. Interestingly, lesions are usually found in bronchial but not pulmonary arteries. Kawasaki disease, on the other hand, is a disease which usually occurs in children and is associated with a desquamating rash which is often accompanied by mucosal ulceration, hence the older terminology “mucocutaneous lymph node syndrome”. Coronary blood vessels are often affected and these children can die of myocardial infarction. The aorta and veins may also be involved. Churg-Strauss syndrome occurs by definition in patients who have a past history of asthma and, in addition to the vasculitic features, these patients demonstrate a profound eosinophilia. The treatment of PAN and Churg-Strauss syndrome has been the subject of a recent review by Guillevin et al and will not be discussed further in this review.24
SMALL VESSEL VASCULITIS
Small vessel vasculitis encompasses Wegener’s granulomatosis, Churg-Strauss syndrome (small and medium sized vessels can be affected), microscopic polyangiitis, Henoch-Schonlein purpura, essential cryoglobulinaemic vasculitis, and cutaneous leukocytoclastic angiitis, the first three of which are strongly associated with ANCA positivity. All these conditions are characterised by inflammation of small to medium sized vessels such as capillaries, venules, arterioles and arteries. Granulomas may (Wegener’s granulomatosis) or may not (microscopic polyangiitis) be found on histological examination of involved tissues. In Wegener’s granulomatosis the granulomatous inflammation must by definition involve the respiratory tract but glomerulonephritis is also common. In Churg-Strauss syndrome the granulomatous inflammatory tissue and the peripheral blood contain a striking number of eosinophils and the patients usually gives a history of asthma which classically precedes the vasculitic illness by up to 20 years. The respiratory tract is frequently affected. In microscopic polyangiitis necrotising glomerulonephritis and pulmonary capillaritis often occur but the upper airways are usually spared. The vasculitis of Henoch-Schonlein purpura is characterised by IgA-dominant immune deposits affecting small vessels of the skin, gut, and glomeruli with arthralgia also being a common presenting feature. Lung involvement in Henoch-Schonlein purpura is rare but pulmonary haemorrhage has been described.25 26 Cryoglobulin immune deposits affecting small vessels of the skin and kidney together with circulating cryoglobulins are the characteristic feature of essential cryoglobulinaemic vasculitis. Isolated cutaneous leukocytoclastic angiitis without evidence of systemic disease or glomerulonephritis is also included in this category.
Disease pathogenesis
Although proteinase-3 is the major autoantigen for c-ANCA which is closely associated with Wegener’s granulomatosis, and myeloperoxidase is the major autoantigenic target of p-ANCA and has been found in patients with microscopic polyangiitis and Wegener’s granulomatosis as well as some disorders which are not overtly vasculitic, other proteinases found within neutrophil granules—such as elastase, cathepsin G, lactoferrin, lysozyme, β-glucuronidase—and in the neutrophil cytoplasm—such as α-enolase—have been reported to be ANCA targets.6 8 It has recently become apparent that ANCA can also target infection related antigens such as bactericidal permeability increasing protein (BPI), human lysosome associated membrane protein 2 (h-lamp-2), defensin, and azurocidin which suggests that ANCA may be associated with various infections. Indeed, the clinical dilemma of ANCA positive vasculitis in association with subacute bacterial endocarditis is clearly established and a recent study by the Cambridge group has shown the presence of BPI ANCA in 80–90% of 66 patients with cystic fibrosis whose titre was highest in those patients with poorest respiratory function.27 None of the 66 patients recognised the classical ANCA targets on ELISA testing although six of these patients had secondary systemic vasculitis and had significantly raised titres of anti-BPI antibodies compared with the remainder. Other workers have also reported the association between cystic fibrosis and systemic vasculitis.28 The importance of standardised assays and further research into target antigens in specific diseases is clear.
The majority of ANCA antigens are microbiocidal components used by the neutrophil in host defence, but what leads to the generation of an immune response to these self antigens is not yet clear and what role infection plays remains to be determined. Certainly, there is evidence that intercurrent infection can lead to an exacerbation of systemic vasculitis29 and that chronic nasal carriage ofStaphylococcus aureus predisposes to relapse in patients with systemic vasculitis.30 There is also convincing evidence that trimethoprim/sulphamethoxazole (Septrin) can play a role in treating limited Wegener’s granulomatosis and in reducing relapse rates in patients with multisystem disease.31-37
Several mechanisms have been put forward to explain how ANCA interacting with target antigens could result in necrotising vasculitis. One theory suggests that neutrophils may be primed by pro-inflammatory stimuli such as cytokines IL-1, TGF-β, TNF, or microbial products which result in translocation of small quantities of ANCA antigens to the surface of neutrophils which then become accessible to ANCA. These cytokines could also lead to the expression and upregulation of adhesion molecules on endothelial cells which is further increased in the presence of ANCA. Furthermore, ANCA have been shown to entice primed neutrophils to release reactive oxygen species and lysosomal enzymes. Adherent neutrophils could thus result in localised endothelial cell injury. These neutrophils may then squeeze between injured endothelial cells and make their way into the perivascular space where they are characteristically seen on histological examination of involved tissues. There is controversy over whether vascular endothelial cells are themselves capable of ANCA antigen expression. Thus, it seems likely that ANCA facilitates neutrophil adherence to vascular endothelial cells and indirectly mediates endothelial cell injury and transmigration of neutrophils into the perivascular space.38-41
Alpha-1-antitrypsin deficiency and cystic fibrosis
Alpha-1-antitrypsin is a major inhibitor of proteinase-3 and deficiency of α1-antitrypsin is associated with a variety of connective tissue diseases including systemic vasculitis. Deficiency phenotypes α1-antitrypsin have been reported in patients with systemic vasculitis. Furthermore, such phenotypes are more common in northern Europeans as is systemic vasculitis.42-44Myeloperoxidase, the other main target of ANCA antibodies in systemic vasculitis, can also inactivate α1-antitrypsin and prolong the activity of proteinase-3. These observations have led to considerable interest in testing patients with known α1-antitrypsin deficiency and significant lung disease for the presence of ANCA. Specks et al 45 have examined serum from 59 patients with α1-antitrypsin deficiency and failed to find a single positive ANCA. A search for α1-antitrypsin deficiency phenotypes in patients with both cytoplasmic and perinuclear ANCA found eight of 40 c-ANCA and two of 40 p-ANCA patients with deficiency alleles, yet measurement of α1-antitrypsin levels were within normal limits in all these patients.46 Although this is theoretically exciting, no clear evidence for a link between α1-antitrypsin deficiency and susceptibility to ANCA positive has been established.
Pulmonary clinical presentation
The lung is the most frequent organ involved in Wegener’s granulomatosis and sometimes, though infrequently (9%), it is the only organ affected. The pulmonary features of Wegener’s granulomatosis have been described by several groups. Most recently, Hoffman et al 47 reviewed the pulmonary findings in 158 patients with Wegener’s granulomatosis followed for between six months and 24 years, a total of 1229 patient years. Although some of the frequencies reported may reflect referral bias as the patients reported were all treated at the National Institute of Allergy and Infectious Disease, USA, nevertheless nasal, sinus, tracheal or ear abnormalities were responsible for initial symptoms in 73% of their patients and occurred at some time in over 90% of patients. Pulmonary infiltrates, nodules, or both were present initially in 45% of patients. Other presenting symptoms included cough (19%), haemoptysis (12%), and pleuritis (10%). During the entire course of illness 66% of all episodes of Wegener’s granulomatosis were associated with either cough (46%), haemoptysis (30%), or pleuritis (28%), and 34% of patients had asymptomatic infiltrates or nodules on chest radiography. Lung disease related to Wegener’s granulomatosis eventually developed in 85% of patients. On the other hand, Cordier et al 48 in a review of 77 patients known to have pulmonary Wegener’s granulomatosis found that 55% of 74 bronchoscopies were macroscopically abnormal with findings varying from bronchial inflammation to stenosis and pulmonary haemorrhage. Seventy two of these patients had pulmonary symptoms ranging from cough (78%) and dyspnoea (56%) to chest pain (32%) and haemoptysis (39%).
TRACHEAL/SUBGLOTTIC STENOSIS
Of the 158 patients reviewed by Hoffman, 25 (16%) had tracheal stenosis which usually presented with shortness of breath. In only five of these patients were the symptoms mild at presentation, and easily responded to treatment. Twenty patients had severe stenoses which were irreversible and 12 patients presented with severe stridor requiring emergency tracheostomy. A total of 17% experienced significant morbidity from pulmonary insufficiency. Somewhat surprisingly, only two of 77 patients described by Cordier et al were found to have subglottic stenosis, although it is not clear from their published work if stenoses producing minor symptoms could have been overlooked.48
PULMONARY HAEMORRHAGE
Haemoptysis has been reported to occur in 12–40% of patients presenting with small vessel vasculitis.49 Others may present with raised carbon monoxide transfer factor or anaemia related to pulmonary haemorrhage or pulmonary infiltrates on chest radiography without overt haemoptysis. While there is a clear association between cigarette smoking and pulmonary haemorrhage in antiglomerular basement membrane antibody-mediated disease, this is not so in small vessel vasculitis. Patients with pulmonary haemorrhage have been reported to have ANCA of the IgM isotype50 51 (most ANCAs are of IgG type although IgA ANCAs have been reported in Henoch-Schonlein purpura). These patients with pulmonary haemorrhage fare badly having a high early mortality from respiratory failure related to the pulmonary haemorrhage or to lung fibrosis after all evidence of pulmonary haemorrhage has resolved.49 In our experience of six recent patients with pulmonary haemorrhage associated either with a new diagnosis or a relapse of small vessel vasculitis, all had IgM ANCA at the time of pulmonary haemorrhage which changed to IgG after the pulmonary haemorrhage resolved. Of these six patients three developed severe respiratory failure approximately one month into treatment when signs of pulmonary haemorrhage had resolved. In two patients bronchoscopic examination yielded evidence of viral infection, RSV, and influenza virus A, respectively. Both patients recovered but one elderly lady died one month after her influenza infection of rapidly progressive respiratory failure which was not due to further pulmonary haemorrhage and no other organism was identified. One patient with pulmonary haemorrhage died shortly after presentation of respiratory failure and circulatory collapse which was complicated by sepsis. Of the four patients remaining alive, three suffer dyspnoea on exertion and have reduced carbon monoxide transfer factor (60–70%) with evidence of pulmonary fibrosis 12–60 months following diagnosis without evidence of current disease activity.
Five per cent of patients with ANCA antibodies have, in addition, circulating anti-GBM autoantibodies which are usually associated with Goodpasture’s disease.52 53 These autoantibodies are known to induce pulmonary haemorrhage in rats exposed to cigarette smoke and are likely to cause pulmonary haemorrhage in humans with the disease. In the case of human Goodpasture’s disease, pulmonary haemorrhage is rare in non-smokers. Turner and Jayne have independently reviewed the clinical picture of patients with both ANCA and anti-GBM autoantibodies and conclude that the pattern of disease in these patients is more like a vasculitis disorder than Goodpasture’s disease.
Pleuropulmonary investigations
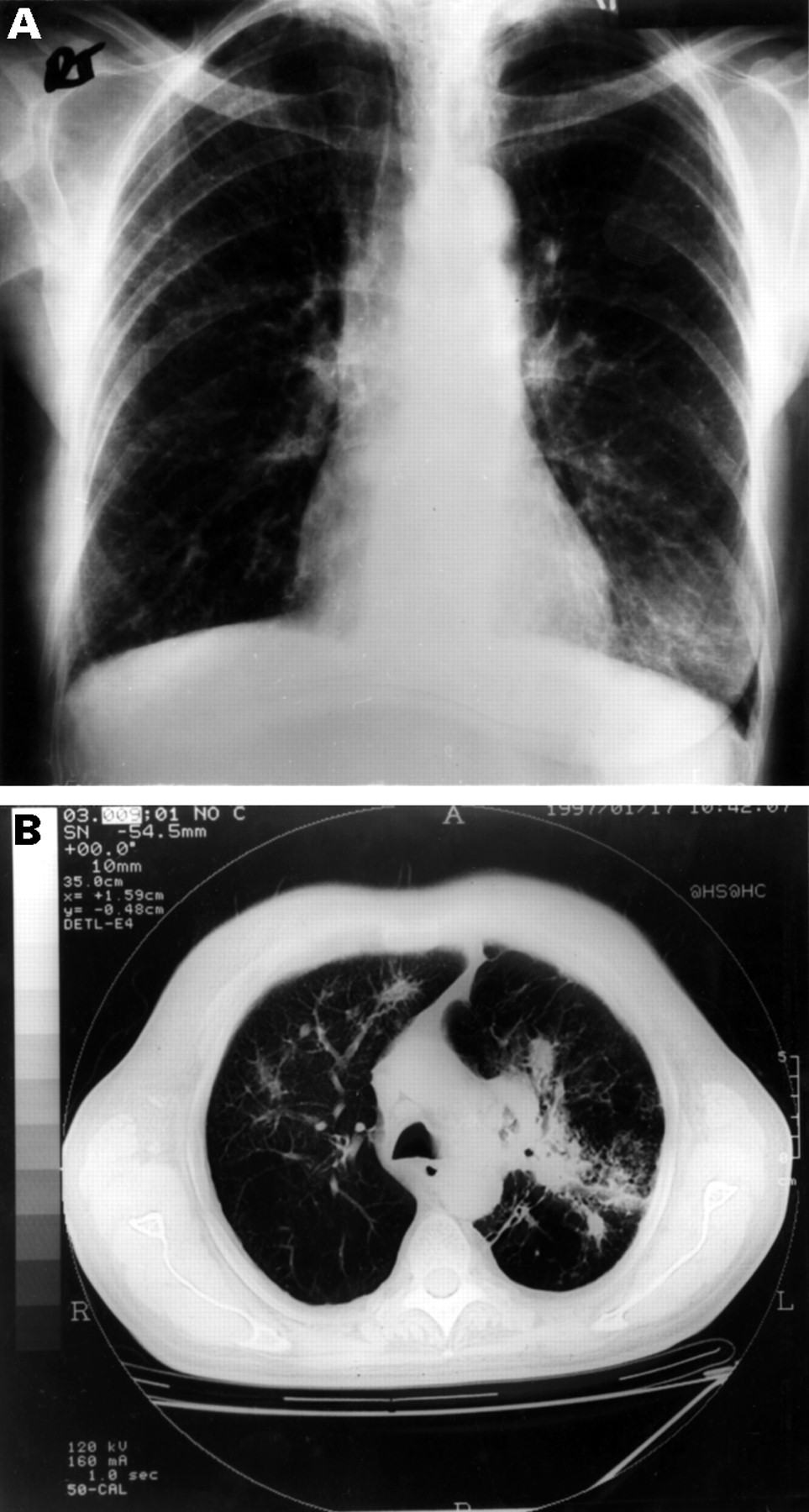
Pulmonary involvement in vasculitis may be identified by an abnormal chest radiograph in an otherwise asymptomatic patient as was the case in 34% of patients studied by Hoffman et al.47 However, in most patients with pulmonary involvement in small vessel vasculitis respiratory symptoms are accompanied by changes which are apparent either on conventional chest radiographs, tomograms, or CT scans. Pulmonary infiltrates (fig 1), nodules (fig 2), or both are the most common finding being present in 45% of patients at diagnosis. The lesions generally involve both sides of the chest although 14 of the 77 patients studied by Cordier et al 48 had unilateral abnormalities. The lung apices are generally spared. The nodules usually consist of rounded lesions with well defined margins varying in size from 5 mm to 100 mm. The largest nodules are usually found to be cavitated (fig 3). Of 53 patients with nodular opacities in Cordier’s study, 26 (49%) were found to have cavity formation.48 CT scanning can undoubtedly pick up nodular and cavitating lesions which are not obvious on chest radiography (fig 4).54 Left untreated, nodules tend to increase in number and to cavitate. Pulmonary infiltrates can have a wide range of appearances in patients with small vessel vasculitis. They may be diffuse, bilateral, low density infiltrates characteristic of pulmonary haemorrhage (fig 1). Patchy, low density infiltrates which can either disappear spontaneously or increase in intensity and dense localised infiltrates which have ill defined margins with occasional air bronchograms are also common. Superinfection of pulmonary lesions is well recognised and may occur as a consequence of treatment.55 56 Purulent bronchorrhoea has been reported either as a consequence of such infection or necrosis of large pulmonary masses. Seven of 41 patients with pulmonary infiltrates in the French series were also found to have cavity formation. Pleural opacities occur in approximately 10% of patients. Other abnormalities such as atelectasis, mediastinal lymphadenopathy, and non-specific calcification have also been reported. Gallium and white cell radionucleotide studies can be useful to identify otherwise silent nasal or pulmonary disease.

(A) Chest radiograph demonstrating diffuse pulmonary infiltrates bilaterally in a p-ANCA positive patient presenting with haemoptysis. (B) Chest radiograph of a c-ANCA positive patient demonstrating pulmonary infiltrates of varying appearance with relative sparing of the apices.

Chest radiograph showing cavitation of a nodule within the left lung together with diffuse pulmonary infiltration and nodule formation in the right lung.

CT scan showing cavitation within a nodule in the right lung.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Chest radiograph and (B) concurrent CT scan from a patient who presented with persistent cough, vasculitic rash, and severe peripheral neuropathy. High titres of anti-myeloperoxidase antibody were detected.
Once a patient has commenced treatment with immunosuppressive agents, interpretation of pulmonary symptoms and chest radiographic findings becomes even more challenging as nearly 50% of patients experience serious infectious complications related to treatment, and almost half of these are due to pulmonary infections with a range of organisms fromPseudomonas, Haemophilus influenzae andPneumocystis carinii to fungal colonisation of existing cavities.55 56 Furthermore, cancer registry data suggest that treated patients with Wegener’s granulomatosis have a 2.4% overall increase in malignancies. While bladder cancers57 58 and lymphoid tumours make up the majority of these, we have seen a patient with small vessel vasculitis in clinical remission who developed what looked like a recurrence of a nodular lesion on chest radiography which turned out to be a metastatic tumour.
Histology
Whether or not it is necessary to obtain pulmonary tissue for histological examination in patients with small vessel vasculitis is controversial.
Hoffman et al 47 performed a total of 82 open lung biopsies in patients with small vessel vasculitis of which 89% showed evidence of combined vasculitis and necrosis. Granulomas and necrosis were found in 90%. Fifty nine transbronchial biopsies were performed in 48 patients; in only four specimens was vasculitis identified and in only three of these were granulomas additionally identified. Thus, the role of transbronchial biopsies in these patients is limited and open lung biopsies are more informative but carry a higher morbidity and mortality. Thus some clinicians recommend that treatment should be initiated in patients with positive ANCA and active clinical symptoms compatible with small vessel vasculitis without histological confirmation of lung involvement. If there is renal involvement a kidney biopsy can be performed with relative ease and the characteristic features of renal vasculitis can confirm the diagnosis and make a lung biopsy unnecessary.
Management of pulmonary involvement in systemic vasculitis
The mainstay of treatment in systemic vasculitis is immunosuppression with steroids and cyclophosphamide.59-64 Other immunosuppressant agents are less effective, especially when disease is generalised. There is a role for Septrin in the management of disease which is limited to the upper airways but stronger therapy is needed if there is evidence of multisystem disease.32-37 Whether this effect is due to immunosuppressant actions of Septrin or to prevention of intercurrent infections which often precede the onset of the initial illness and can provoke relapses is not yet clear.29 There is evidence, though not as yet tested in randomised trials, for a beneficial effect of intravenous immunoglobulin (IVIG) treatment,65 yet the ability of IVIG to remove circulating ANCA antibodies varies considerably depending on the batch of immunoglobulin used and on the manufacturer, which reflects the difficulty in quantifying ANCA specific anti-idiotype antibody levels in individual batches of donor serum. Similarly, concern regarding the possibility of viral transmission using such products is justified. Plasma exchange has been shown in a randomised trial of vasculitis with renal involvement to benefit patients whose presenting creatinine level is above 500 mmol/l.66 There is a theoretical advantage to plasma exchange in patients with pulmonary haemorrhage who have circulating IgM ANCA antibodies which can be successfully removed using a centrifuge plasma exchange technique rather than a plasma filter. However, there are currently no randomised trials to validate this view; furthermore, plasma exchange also removes circulating fibrinogen and thus, in theory, could worsen pulmonary haemorrhage. This potential problem can be overcome by exchanging patient’s plasma for fresh frozen plasma in those at risk of haemorrhage. Blood transfusion and ventilatory support may also be necessary in these patients and careful searches for superadded infection with early and appropriate therapy is mandatory. Antithymocyte globulin and monoclonal antibodies designed to remove circulating T cells have also been used in refractory cases.67-69
The optimum duration of treatment is again a controversial subject. More patients are currently lost from over-aggressive treatment leading to infection than from failure of treatment to control disease. Similarly, there is much concern over the long term consequences of immunosuppression, particularly bladder toxicity and malignancy with cyclophosphamide usage.57 58 Most UK units now use cyclophosphamide in doses of 2–3 mg/kg per day orally, together with tapering doses of prednisolone starting at 60 mg per day for the first week followed by 45 mg for the second week and 30 mg for the third week, after which time steroids are reduced by 5 mg per day each week if the patient’s response continues to be favourable. This therapy should be combined with H2 blockers or proton pump inhibitors to help prevent gastric and duodenal ulceration. Prevention of fungal infections by the routine use of suspensions and lozenges is also recommended. Septrin prophylaxis for the prevention ofPneumocystis carinii infection is widely practised during the first six months of treatment and may, of course, have additional beneficial effects as discussed above. In most units it is customary to switch patients from the more toxic agent cyclophosphamide to azathioprine or, recently, the newer agent, mycophenylate mofotil,70 71 once disease remission has been achieved and sustained for three months. However, whether this switch is best carried out at three or 12 months is the basis of an ongoing randomised trial, the results of which are eagerly awaited. Cyclophosphamide doses should be tailored to maintain the white cell count above 4 × 109/dl. Other immunosuppressant agents such as chlorambucil, methotrexate, cyclosporin, and azathioprine alone are less commonly used and are generally less effective.
The role of ANCA testing in optimising treatment for individual patients is gradually becoming clear. Certainly, it is now recognised that persistently ANCA positive patients are at greatest risk of disease relapse. Patients who are intermittently ANCA positive generally seroconvert before a clinical relapse. Reduction and withdrawal of treatment from patients who remain ANCA negative after initial treatment is now widely practised and appears to be safe, although regular monitoring with ANCA is advised as patients may relapse decades after initial presentation. More recently, some workers have been using titres of ANCA antibodies as determined by solid phase assays to guide treatment alterations.13 There is some evidence to support this practice but the general principle of treating patients rather than the test should still be upheld.
Outcome
Patient survival has improved dramatically over the last four decades since the original descriptions by Wegener, all of whose patients died within seven months of diagnosis.72 73Walton reported 80% mortality by the end of the first year, rising to 90% by the end of the second year in his 1958 series.74 A more recent series of 151 patients who received treatment at the Mayo clinic reported a 10% mortality at the end of one year which rose to 24% by the end of five years of follow up.59 In this series De Remee et al also recognised that one quarter of their patient deaths (12 of 43) occurred because of opportunistic lung infections resulting from the use of immunosuppressive drugs. Others groups have reported similar results.55 56 The most ominous manifestation of disease in the lungs is alveolar haemorrhage which has been associated with high early mortality.49However, pulmonary haemorrhage may have more long term consequences such as pulmonary fibrosis. Pulmonary opacities not due to pulmonary haemorrhage usually disappear within weeks of commencing treatment with immunosuppressant drugs, parallelling improvement in other organs. The great challenge of the future remains to design treatment regimens which achieve maximum disease control while minimising complications associated with the treatment.